 US
US IN
INBy MedGenome Scientific Affairs
Overview of Transcriptomics
Transcriptome sequencing/RNA sequencing allows unbiased characterization of global gene expression profiles associated with different cells/tissues. As genes govern cellular function, transcriptome profile can provide valuable insights into molecular mechanisms operating in a biospecimen. RNA sequencing has transformed biological research by discovering almost all transcripts encoded by a genome including mRNAs, long non-coding RNAs and miRNAs. It has also revealed many alternatively spliced variants which is a common feature among complex multicellular organisms. It has revolutionized biomedical research by enabling characterization of global gene expression profiles associated with cells/tissues in healthy and disease conditions. Understanding molecular mechanisms of disease has paved way for identification of biomarkers and development of novel drugs targeting specific genes that drive disease pathology.
RNA sequencing has been widely used to study tumor microenvironment and the interactions between cancer cells and immune cells, providing insights into mechanisms of tumor immune evasion and potential targets for immunotherapy. In infectious disease research, RNA sequencing has been used to study host-pathogen interactions and identify host factors that contribute to disease susceptibility or resistance. By analyzing gene expression profiles of infected cells, researchers have uncovered molecular mechanisms underlying pathogen replication and host immune responses. This knowledge can aid in the development of new antiviral therapies and vaccines.
Applications of RNA Sequencing
-
- Gene expression profiling of cells/tissues
- Differential gene expression profiling to identify and quantify gene expression differences between healthy and disease tissues
- Identification and characterization of alternatively spliced transcripts
- Identification of biomarkers based on gene expression signatures associated with different diseases
- Identification of drug targets based on molecular mechanisms that drive disease pathology
- Identification of oncogenic fusion transcripts that drive cancers
- Characterization of molecular mechanisms associated with host-pathogen interactions in infectious diseases
- Characterization of gene expression at single cell resolution
MedGenome offers a variety of RNA sequencing services based on sample quality and amount of available starting material.
Types of RNA sequencing services offered by MedGenome
| Library Prep Services | Assay Type | Stranded Type | Starting Material | Input Amount |
|---|---|---|---|---|
| TruSeq Stranded mRNA | mRNA Seq | Yes | RNA | 100 ng – 1 μg RNA |
| Illumina Stranded mRNA | mRNA Seq | Yes | RNA | 25 ng – 1 μg RNA |
| Takara SMART-Seq V4 | mRNA Seq | No | RNA, Cells | 10 pg – 10 ng RNA, 1 – 1,000 cells |
| TruSeq Stranded Total RNA | Total RNA | Yes | RNA | 100 ng – 1 μg RNA |
| Pico V2 / V3 | Total RNA | Yes | RNA | 250 pg – 10 ng RNA |
| SMART-Seq Stranded | Total RNA | Yes | RNA, Cells | 10 pg – 10 ng RNA, 1 – 1,000 cells |
Single-Cell RNA Sequencing
Distinct gene expression profiles are associated with different cell types. Single-cell RNA sequencing has emerged as a powerful tool, allowing researchers to unravel the heterogeneity and complexity of biological systems. This technique enables the identification and characterization of rare cell populations, which are often missed in bulk RNA sequencing. By analyzing gene expression profiles at the single-cell level, researchers can identify cell types, define cell states, and uncover novel disease-associated biomarkers. The same technology is now widely used for characterizing immune repertoire by sequencing T-cell and B-cell receptors. It has also enabled identification of heavy and light chain pairs from B cells to characterize antigen specific antibodies.
The process of single-cell RNA sequencing involves multiple steps, including cell isolation, RNA extraction, library preparation, sequencing, and data analysis. Cells are typically dissociated from the tissue of interest and captured in microfluidic devices or droplet-based systems. Individual cells are then lysed, and RNA molecules are extracted and converted into complementary DNA (cDNA). The cDNA is amplified, and sequencing libraries are prepared for high-throughput sequencing.
Data analysis in single-cell RNA sequencing is a complex task due to the large number of cells and the high-dimensional nature of the data. Bioinformatic tools and algorithms are used to cluster cells based on their gene expression profiles, identify differentially expressed genes, and infer cellular trajectories. By integrating single-cell RNA sequencing data with other omics data, such as genomics and proteomics, researchers can gain a more comprehensive understanding of disease mechanisms. MedGenome uses 10x Genomics platform for offering single cell transcriptomics services.
Data Generation and Analysis
RNA sequencing generates vast amounts of data, which require sophisticated computational tools and algorithms for analysis. The raw sequencing data undergoes quality control to remove low-quality reads and sequencing artifacts. The processed reads are then aligned to a reference genome or transcriptome to determine the origin of each read.
Once the reads are aligned, the next step is to quantify gene expression levels. This involves counting the number of reads that map to each gene or transcript. Various statistical methods are used to normalize the expression data and identify differentially expressed genes between different conditions or cell types.
Data analysis in RNA sequencing also involves the identification of alternative splicing events, non-coding RNAs, and fusion genes. These events can provide valuable insights into disease mechanisms and potential therapeutic targets. Furthermore, RNA sequencing data can be integrated with other types of omics data, such as genomic and proteomic data, to unravel complex interactions and regulatory networks.
Looking Ahead: The Future of RNA Sequencing
The field of RNA sequencing is rapidly evolving, with new technologies and analytical approaches being developed. One of the major challenges in RNA sequencing is the analysis of low-quality or degraded RNA samples. Researchers are actively working on improving the sensitivity and accuracy of RNA sequencing methods to overcome this limitation.
Another area of active research is the integration of RNA sequencing data with other omics data, such as genomics, proteomics, and metabolomics. Integrative omics analysis can provide a more comprehensive understanding of disease mechanisms and identify novel therapeutic targets. Machine learning and artificial intelligence algorithms are being developed to analyze and interpret large-scale omics data, enabling the discovery of complex interactions.
In conclusion, RNA sequencing has revolutionized biomedical research by providing a comprehensive view of gene expression patterns and molecular signatures. It has enabled the identification of biomarkers for various diseases and has shed light on the underlying mechanisms of complex diseases. With continuous advancements in technology and data analysis, RNA sequencing holds great promise for personalized medicine and the development of targeted therapies.
MedGenome RNA Solutions or Case study
Papillary thyroid carcinoma (PTC) is one of the most common forms of thyroid cancer with >90% of cases achieving remission post-surgery. Despite this favorable outcome, the emergence of aggressive variants underscores the growing need for personalized therapeutic approaches.
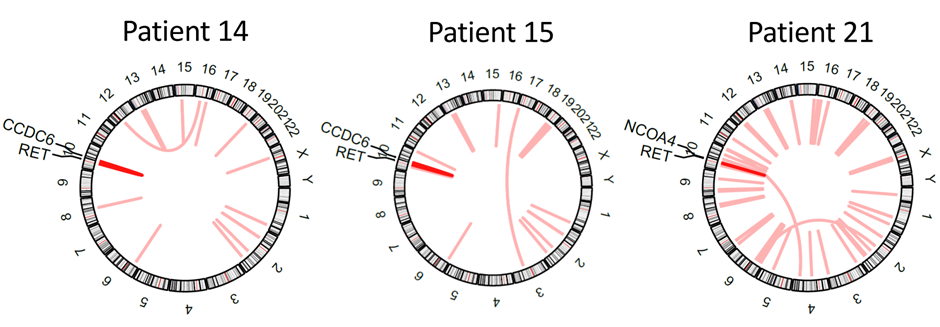
In collaboration with researchers at the University of Mainz, Germany, MedGenome generated RNA sequencing and proteomic data from PTC tumor samples from 22 patients1. Multiomic analysis identified a novel rearrangement in one of the patients. This novel rearrangement led to a BAIAP2L1-BRAF fusion gene product that transformed immortalized human thyroid cells. We also identified two previously known RET fusions in two other patients (Figure 1) as well as other druggable targets including TRIM25, PKCδ, and PDE5A.
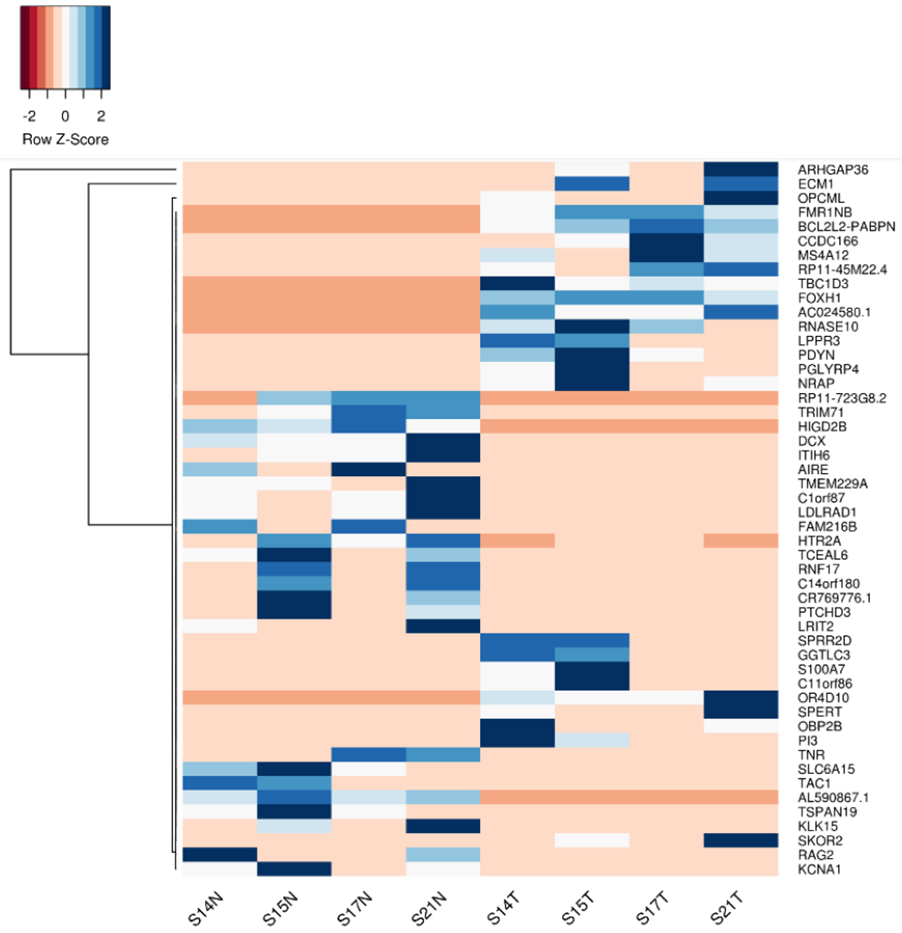
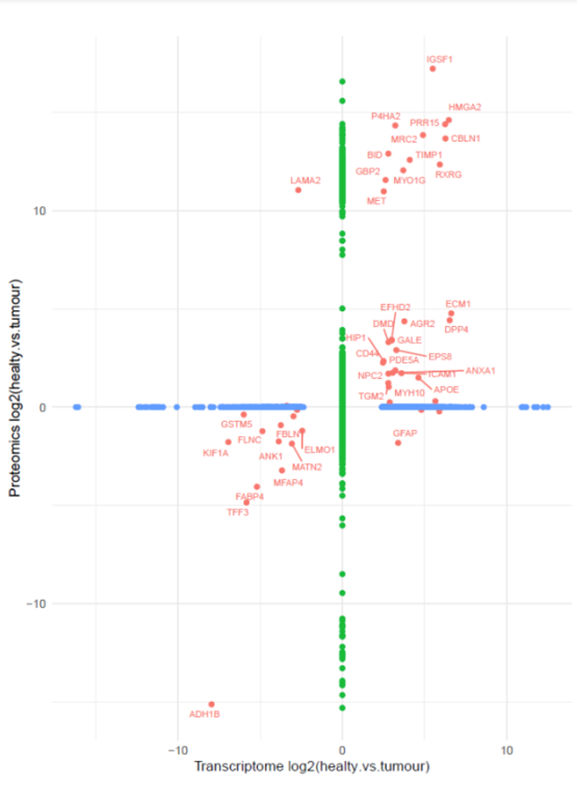
Integrative analysis of RNA-seq and proteomics data was performed using the list of differentially expressed genes (Figure 2) and proteins derived independently from RNA-seq and proteomics data, respectively in the fusion-carrying patients to identify factors significantly deregulated at both the mRNA and protein levels. This analysis yielded 20 identified factors (Figure 3), including PDE5A and IGSF1 (also called p120), a factor known to be associated with hypothyroidism, which are upregulated in tumor and/or metastatic tissue in comparison to the matching normal tissue.
Taken together, this study demonstrates the power of multiomic analyses to identify and characterize cancer therapy targets, which in turn can advance precision medicine and personalized therapeutics.
If you are interested in learning more about RNA sequencing and its applications in disease research connect with our scientific team by writing to us at research@medgenome.com
References
-
- Renaud, E., Riegel, K., Romero, R. et al. Multiomic analysis of papillary thyroid cancers identifies BAIAP2L1-BRAF fusion and requirement of TRIM25, PDE5A and PKCδ for tumorigenesis. Mol Cancer 21, 195 (2022).
#Transcriptome sequencing, #RNA Sequencing, #Biomarker Identification, #Gene Expression, #Spliced transcripts, #Single cell resolution, #Single-cell RNA sequencing
