 US
US IN
INBy Parimala Nagaraja, Scientist, NGS, MedGenome Inc.
NGS technologies is at the forefront of Biological Research. They produce enormous data running into gigabases in a single round of sequencing. However, several sequencing artifacts such as read errors (base calling errors and small insertions/deletions), poor quality reads and primer/adaptor contamination are quite common with the NGS data obtained after sequencing. It can impose significant impact on the downstream analysis such as sequence assembly, single nucleotide polymorphisms (SNP) identification and gene expression studies.
Quality control metrics play a critical role in ensuring to minimise the number of errors and help in achieving high quality data for a successful experimental study. MedGenome strives to maintain strict guidelines in terms of QC metrics to achieve high quality data for our clientele.
QC metrics are mainly applied at 3 levels:
- • Sample QC (DNA/RNA)
- • Library QC
- • Sequencing QC
Sample QC
An Ideal NGS assay would require high quality DNA/RNA which is usually determined using Tapestation/Bioanalyzer that provide the DIN/RIN (DNA/RNA Integrity number) values ranging between 1-10, where 10 is the highest quality sample and 1 is the highly degraded and poor-quality samples.
Depending on the assay type and Sample source, MedGenome has a set of guidelines in terms of Quantity, Quality and Volumes for the clients. At MedGenome, all samples are first subjected to QC using Qubit to determine the quantity and Tapestation/Bioanalyzer to determine the quality.
Based on the QC determined, samples are classified as a Pass or Marginal or Fail. Replacement samples are usually requested for the samples that failed Sample QC. For Marginal samples, replacements are highly encouraged, else they will be proceeded to library preparation after client’s approval.
Library QC
All libraries which are prepared in-house are checked for their quality using Tapestation/Bioanalyzer and quantified using Qubit. Tapestation and Bioanalyzer results are thoroughly reviewed for the expected library size, adapter contamination, primer dimers and PCR artifacts before they are pooled and loaded onto the sequencer. MedGenome also offers sequencing support for Premade libraries which are prepared by various clientele based on their project requirements. All premade libraries are also subjected to MedGenome QC methodologies and are diligently reviewed and classified as Pass or Marginal or Fail before sequencing. Following Images provides an example for Good vs Bad Library QC.
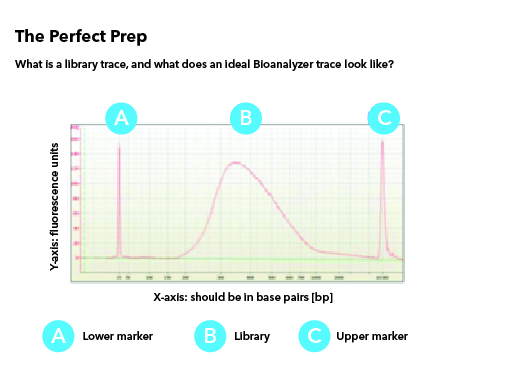
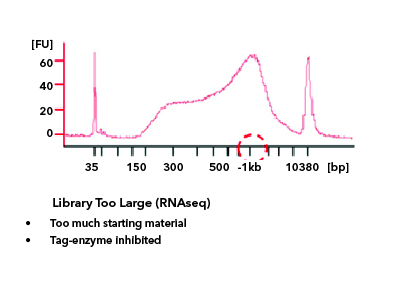
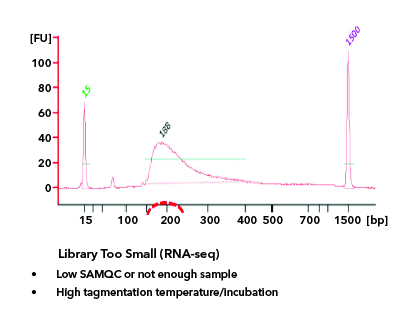
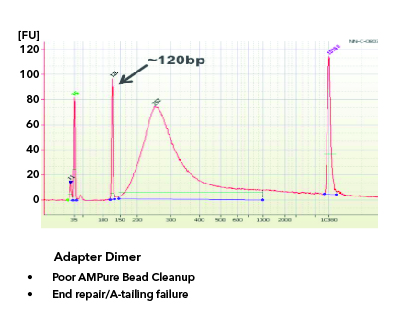
Sequencing QC
Illumina facilitates the users to monitor the runs in real time without interfering with the run performance using a software called Sequencing Analysis Viewer (SAV). This software is compatible with all HiSeq, NextSeq, MiSeq and NovaSeq platforms. The following table describes the features used for evaluating the Sequencing QC:
Table 1: Different terms and their corresponding definitions as viewed in SAV.
| Term | Definition |
|---|---|
| Intensity | The 90% percentile extracted intensity for a given image (lane/tile/cycle/channel combination). On platforms using four-channel sequencing, 4 channels (A, C, G, and T) are shown. |
| FWHM | The average full width of clusters at half maximum (representing their approximate size in pixels). |
| % Base | The percentage of clusters for which the selected base has been called. |
| %Q >/= 20, %Q >/=30 | The percentage of bases with a Phred or Q quality score of 20 or 30 or higher, respectively |
| Density | The density of clusters for each tile (in thousands per mm2). |
| Density PF | The density of clusters passing filter for each tile (in thousands per mm2). |
| Clusters | The number of clusters for each tile (in millions). |
| Clusters PF | The number of clusters passing filter for each tile (in millions. (Metrics given in below images) |
| % Pass Filter | The percentage of clusters passing the Chastity filter (Metrics given in below images) |
| % Phasing, % Prephasing | The average rate (percentage per cycle) at which molecules in a cluster fall behind (phasing) or jump ahead (prephasing) during the run. |
| % Aligned | The percentage of the passing filter clusters that aligned to the PhiX genome. |
| Error rate | The calculated error rate, as determined by the PhiX alignment. Subsequent columns display the error rate for cycles 1–35, 1–75, and 1–100. |
| Yield Total | The number of bases sequenced, which is updated as the run Progresses. (Metrics given in below images) |
| Projected Total Yield | The projected number of bases expected to be sequenced at the end of the run. |
Illumina provides the standardised expectations of reads outputs, Reads Passing Filters, and Quality Scores for each Flow Cell type on every sequencing platform. Following Images provide the metrics for different flowcells on NovaSeq 6000.

Image Source: https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.html

Image Source: https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.html
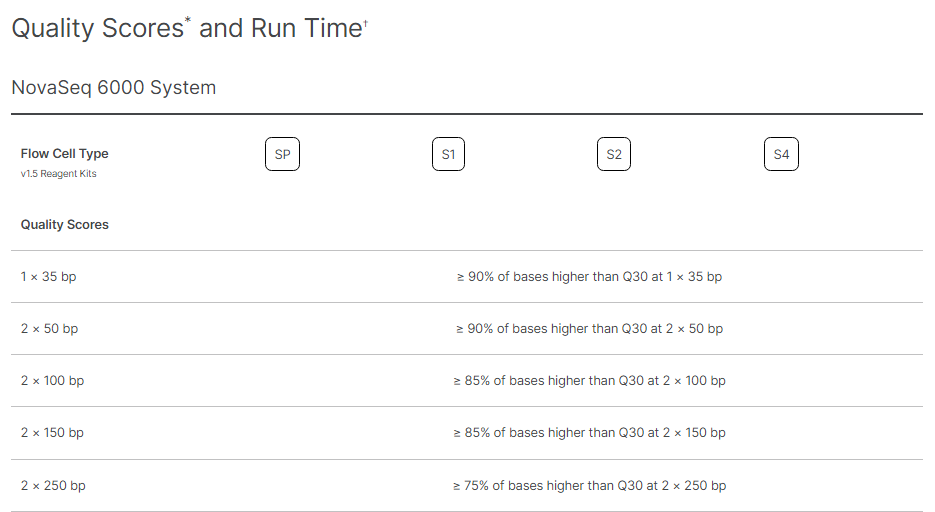
Image Source: https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.html
Sequencing QC also depends on the library types pooled into the same lane or Flow Cell. If libraries prepared using the same protocol (For ex: Illumina Stranded mRNA) are pooled and sequenced, we can see NovaSeq outperforming the Illumina specifications. However, this is usually not the case in an ideal world for any NGS service providing company with high throughput fast paced Turn Around Times. Hence, when multiple libraries of different library types are pooled, it is expected to see the variations in the run performances and the data yields. Following images provide an example of the Sequencing stats achieved by pooling similar libraries and Mixed libraries.
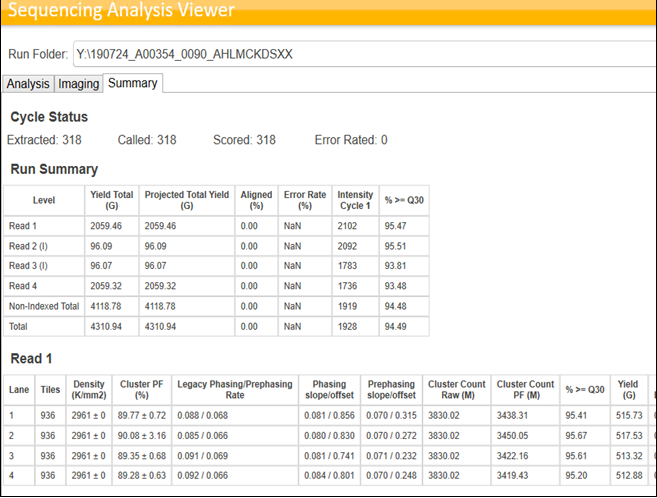
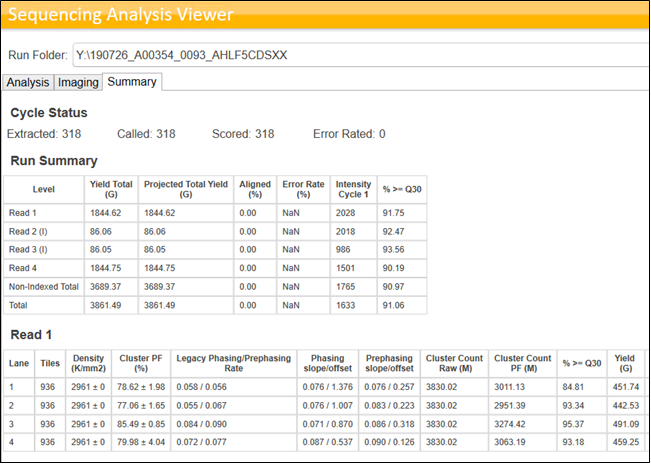
Quality Control of the Sequencing raw data
Raw data quality control should be the initial step of data analysis for any successful study. There are several tools that are publicly available for conducting quality control on raw FASTQ files. FastQC developed by Babraham Institute bioinformatics group is one of the most popular tools that offers QC control parameters such as average base quality score per read, the GC content distribution and identification of the most duplicated reads.
The important parameters to check for raw sequencing data quality are:
- • Base Quality
- • Nucleotide distribution
- • %GC distribution
- • PCR duplicates
Base Quality check:
A common way to visualize base quality is to draw a base Q-score versus cycle plot.
Sequencing data generated on Illumina platforms tend to observe a median base quality score between 35 and 40 in the Phred scale. Large variations in base quality scores (Figure 10a) usually indicate poor Library QC. Sudden drop in the Quality scores (Figure 10b) usually indicate Adapter dimer contaminations or Fluidics issue in the instrument. For paired-end reads, it is common to observe higher quality in the first end of the read than the second end owing to the amount of time the template was on the instrument and increasing laser exposure over time.
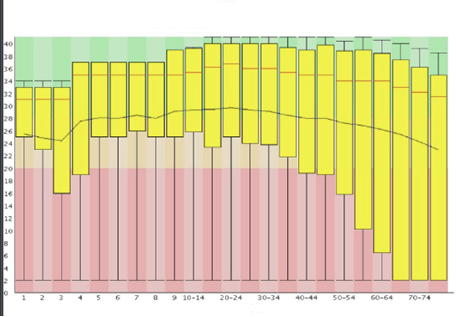
Image Source: Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2014 Nov;15(6):879-89. doi: 10.1093/bib/bbt069. Epub 2013 Sep 24. PMID: 24067931; PMCID: PMC4492405.
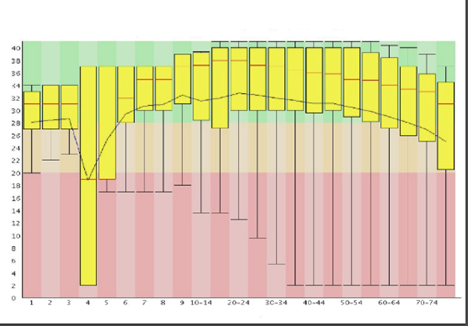
Image Source: Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2014 Nov;15(6):879-89. doi: 10.1093/bib/bbt069. Epub 2013 Sep 24. PMID: 24067931; PMCID: PMC4492405.
Nucleotide Distribution
This parameter is useful for Whole genome and Whole exome libraries (High diversity) but not for Amplicons or RNA libraries (Medium-Low diversity). For a perfect sequencing run, the distribution of the four nucleotides (A T C G) across all reads should remain relatively stable (Figure 11)
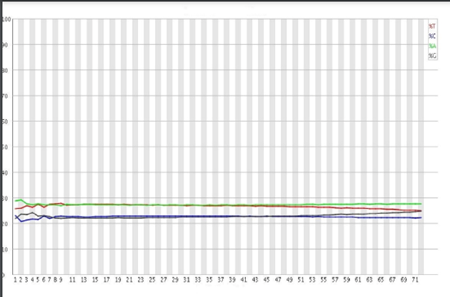
Image Source: Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2014 Nov;15(6):879-89. doi: 10.1093/bib/bbt069. Epub 2013 Sep 24. PMID: 24067931; PMCID: PMC4492405.
%GC Distribution
The percentage of GC in the genome varies across species and across the regions of each genome. For exome regions, the GC content is about 49–51%, while for whole-genome sequencing (Human), the GC content is around 38–40%. Abnormal GC content percentage (>10% deviation from normal range), can indicate contamination.
PCR Duplicates
PCR duplicates arise during library preparation when PCR amplifies the fragments with adapters. Presence of PCR duplicates can lead to potential biases in variant calling algorithms. Hence these are removed by most of the Bioinformatic analysis pipelines during the pre-processing of the data. General causes for high rate of PCR duplicates are Low input quantity, Over sequencing, too many PCR cycles, Low pre-enrichment yield/final library yield, and short library fragments.
Conclusion
MedGenome strives to follow all the best practices in Lab and QC methodologies. Apart from just performing QC, we also interpret and communicate with the client regarding any deviations from MedGenome’s QC standards and recommend the best possible actions to proceed. After the sequencing is performed to the best of our abilities, the raw data is thoroughly reviewed as per Illumina’s standards prior to the data being shared with clients. MedGenome also offers data and sample storage facilities as per clients’ requests.
References
-
-
- 1. https://horizondiscovery.com/en/blog/2020/the-5-ngs-qc-metrics-you-should-know
- 2. https://support.illumina.com/content/dam/illumina-support/documents/documentation/software_documentation/sav/sequencing-analysis-viewer-user-guide-15020619-f.pdf
- 3. https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.html
- 4. https://www.frontiersin.org/articles/10.3389/fgene.2014.00111/full
- 5. Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2014 Nov;15(6):879-89. doi: 10.1093/bib/bbt069. Epub 2013 Sep 24. PMID: 24067931; PMCID: PMC4492405.
-
#NGSQC, #QCmetrics, #readquality, #sequencingQC, #LibraryQC, #DensityPF, #ClustersPF, # %PassFilter, #Errorrate, #YieldTotal
