By Savita Jayaram Ph.D., Kushal Suryamohan Ph.D., MedGenome Scientific Affairs
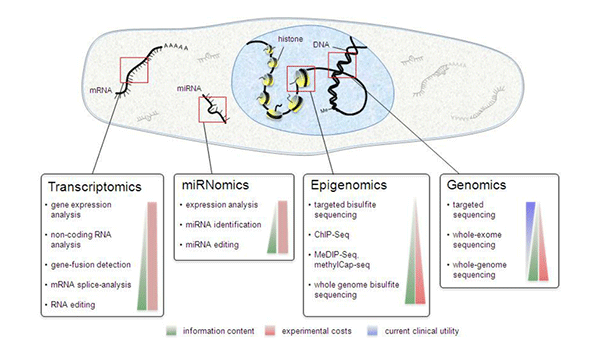
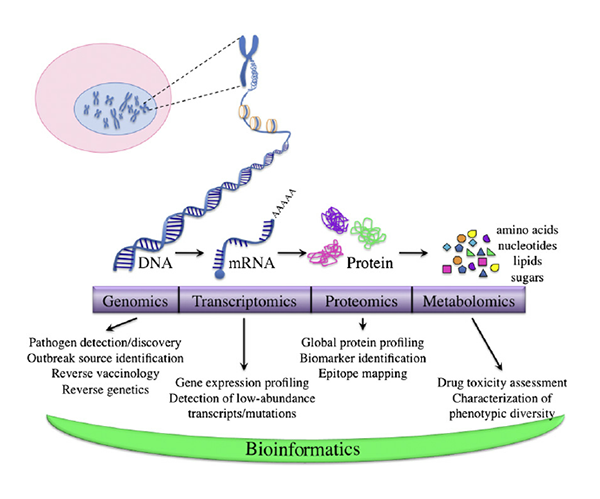
Recent advances in next-generation sequencing technologies have heralded a paradigm shift in the field of precision oncology and personalized/genomic medicine, with a large number of somatic- and germline mutation-profiling programs worldwide. These programs have paved the way for personalized medicine in contrast to a unified approach that clearly fails in select individuals, conferring benefits to only a subset of patients. While these genomic analyses become increasingly accessible and almost commonplace to all research scientists, clinicians and molecular geneticists, they are faced with the challenging task of interpreting and translating the results from these analyses.
Owing to the inherent heterogeneity and complexity of solid and liquid tumors and inherited cancers, newer bioinformatics tools are required to interpret and prioritize the variants, including SNPs, In-Dels, copy number variations (CNVs), translocations, gene fusions, and splice variants, covering a broad spectrum of genomic alterations. Recently, MedGenome Labs launched the AI-enabled VarMiner pipeline to detect actionable genetic variants in rare and inherited cancers, powered by internally benchmarked tools and databases, to precisely pinpoint these changes with accuracy and efficiency. Identifying these causal variants is like pulling out a needle in a haystack, but is the need of the hour, to improve prediction rate with higher specificity.
Custom Tumor Panels
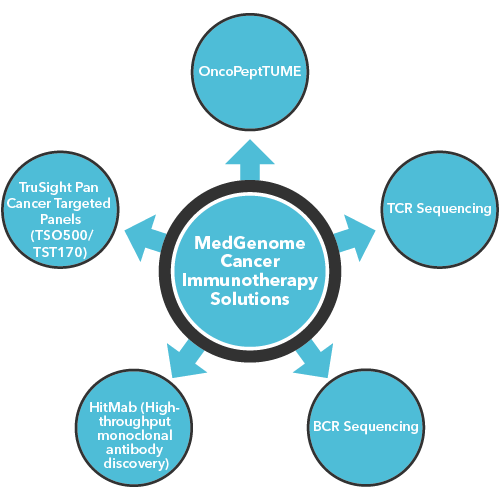
Further, to provide an easy identification and maximize the utility of these analyses, MedGenome has developed many tumor panels such as TSO170 and TST500. These panels enable fast and seamless reporting ranging from blood-based markers from circulating tumor cells (from liquid biopsies) to high depth sequencing of tumor mutation burden to provide insights into the potential solutions for the patients, in both research and clinical settings. TruSight Oncology 500 offers wide variety of benefits in analyzing multiple tumor variants across 523 genes in a single assay, enabling comprehensive genomic profiling of tumor samples. The assay is highly effective in identifying all types of relevant DNA and RNA variants in different types of solid tumors encompassing sarcomas, lung, melanoma, ovarian, breast, gastric, and bladder cancers. Also, the assay is highly accurate in measuring immuno-oncology biomarkers such as microsatellite instability (MSI) and tumor mutational burden (TMB). Our TST170 Panel had been validated on circulating tumor DNA (ctDNA) providing an in-depth view into cancer genetics.
Neo-antigen prediction
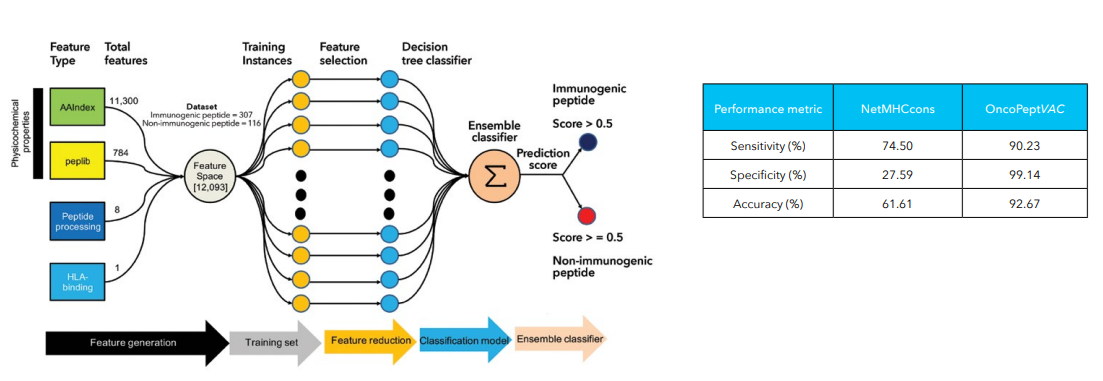
OncoPeptVACTM
One of the greatest achievements in cancer therapies in the past decade has been the introduction of immunotherapy drugs such as Nivolumab and Ipilimumab targeting immune checkpoint inhibitors, PD1 or PDL1 and/or CTLA4, significantly improving clinical outcomes. Notwithstanding a high overall response rate to these drugs, long-term benefit is realized by only a small fraction of the treated patients. Additionally, a potential downside of these antibody drugs such as bispecific antibodies, and chimeric antigen receptor [CAR]-T cells is they can themselves elicit potential immunogenicity effects inducing anti-drug antibodies, on treatment.1 This led to the advent of personalized neoantigen-based cancer therapies and adoptive T cell therapies, that have been shown to prime host immunity against cancer. Despite their growing popularity, cancer vaccines have only had modest success. One of the key impediments to the development of effective cancer vaccines has been the difficulty to select ideal neoantigen candidates. Neoantigens are predicted by exploiting tumor-specific mutations derived from gene fusions, frameshifts, splice variants or other aberrations that sufficiently distinguish it from self-antigens. Neo-antigen prediction helps to identify such 9-15mer neoepitopes candidates for vaccine development, that can elicit a strong disease-specific immunogenic response. Several studies have shown that among the immunodominant epitopes identified for influenza, HIV, SARS-CoV-2 and so on, only a handful of them induced a strong cytolytic CD8 and/or CD4 T-cell response. This necessitated the development of tool that could accurately predict ideal neoantigen candidates for immunotherapy that can be tested, with further broader applications in oncology therapeutics.
We developed a novel proprietary and now patented algorithm, OncoPeptVACTM that can not only accelerate identification of cancer vaccine candidates but also identify immunogenicity risks of antibody-based drugs.2 The algorithm driven by machine learning approaches incorporates features associated with presentation of the antigen on the surface and utilizes features regulating T cell receptor (TCR) binding of the HLA-peptide complex assigning accurate prediction scores for neo-epitope prioritization and neo-antigen prediction. OncoPeptVACTM identifies immunogenic peptides from exome as well as RNA-seq data from tumor/normal pairs, to predict CD8 T-cell activating epitopes. This pipeline was successfully validated in two different studies. Following neoepitope prioritization using OncoPeptVACTM pipeline, three mutant peptide antigens were selected from Lynch syndrome-colorectal cancer patients and shown to induce a potent CD8 T cell response.3 In another recent study, immunodominant T-cell epitopes of SARS-CoV-2 spike antigens showed robust pre-existing T-cell immunity in unexposed individuals, contributed by TCRs that recognize common viral antigens such as influenza and CMV.4 Interestingly, these viral epitopes lacked sequence identity to the SARS-CoV-2 epitopes. Both studies were published in Nature, Scientific Reports. Further ongoing studies from MedGenome showed the effects of peptide length and peptide dosage on CD8 T-cell activation. The immune response of a 9mer or 15mer version of HLA-2-restricted ‘GILGFVFTL’ epitope was compared to determine which made a better vaccine candidate, by measuring the CDR3 expansion as a measure of T-cell epitope engagement diversity.5 It was seen that the 15mer epitope produced a more robust and sustained response, and private CDR3s not expanded by 9mer peptides. All these studies, show the potential utility of our pipeline in accurately predicting prototypical immunodominant vaccine candidates that can be further screened using our proprietary OncoPeptSCRNTM T-cell assay platform described below.
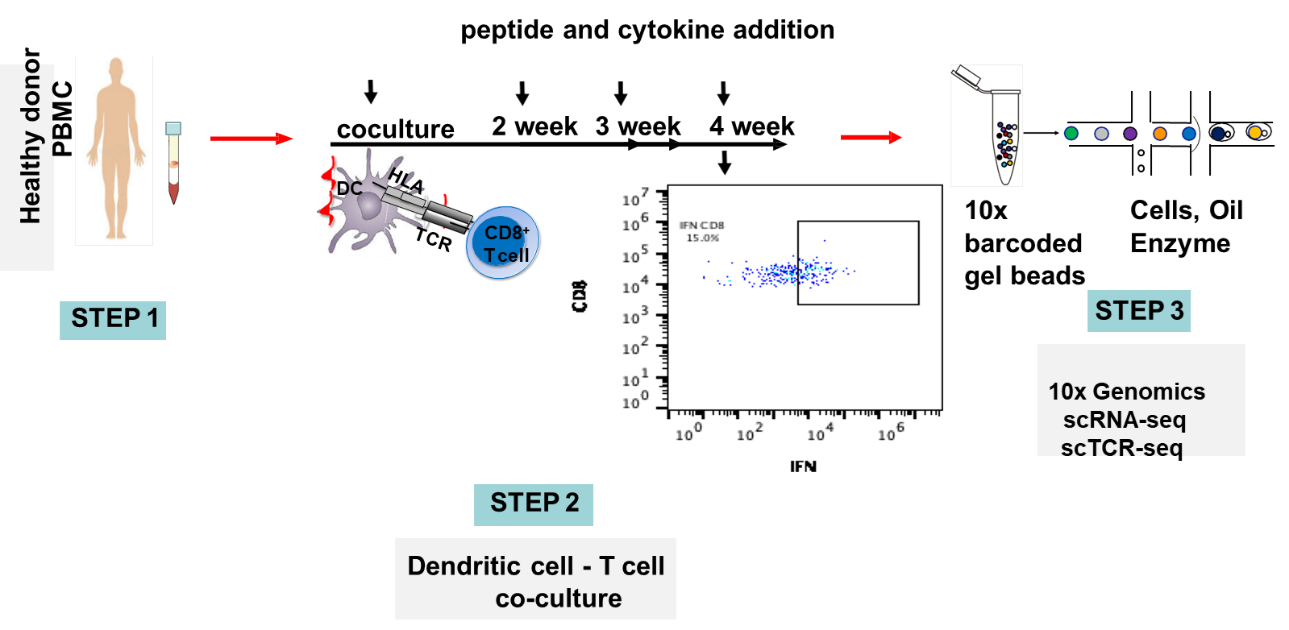
MedGenome’s OncoPeptSCRNTM
Therapeutic revival of tumor-specific exhausted T cells using neutralizing antibodies targeting the immune checkpoint inhibitors, namely, T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) has significantly improved clinical outcomes in cancer. T cells exist in a wide spectrum of functional states – from fully functional at one end of the spectrum, to fully dysfunctional at the other end. One of the factors governing the fate of the tumor response to checkpoint inhibitors is the ratio of functional to dysfunctional state of T cells, which in turn is modulated by a wide array of immune-suppressive signals present within the tumor microenvironment. Tumors that are immunologically ‘hot are characterized by high infiltration of activated T cells that also express PD1 and CTLA4. These inhibitory receptors evolved to prevent over activation of the immune system but cancer cells hijack this mechanism to their benefit by expressing the corresponding ligands driving the T cells to exhaustion.5 Targeting these checkpoint inhibitors can reverse this dysfunctional state and reinvigorate the immune response, only if they are in a ‘partially exhausted’ state. This is the basis for the development of immunotherapy drugs such as Nivolumab(anti-PD1) and Ipilimumab (anti-CTLA4) that can rescue the T cells from exhaustion.
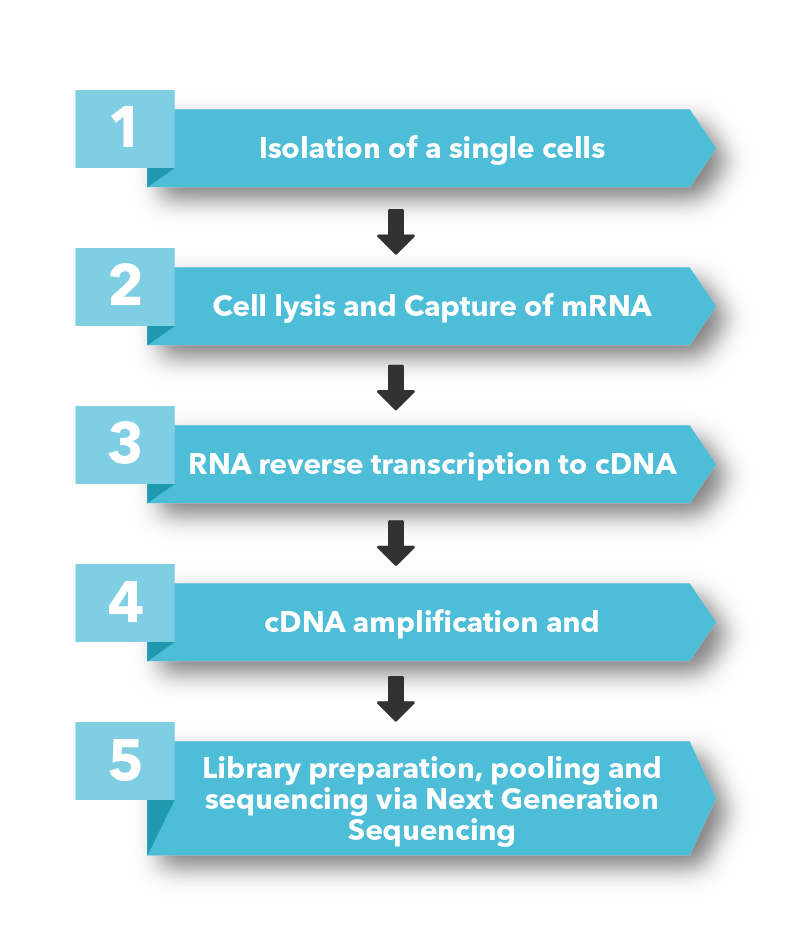
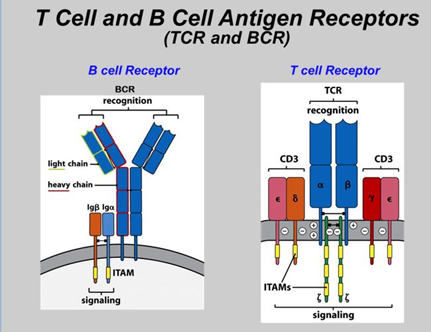
The OncoPeptSCRNTM T-cell Activation Assays leverages the single cell transcriptomics on a 10X Genomics platform to assess immunogenicity of HLA-peptide pairs. The cancer peptides/antigens selected using our OncoPeptVACTM platform are expressed as a minigene or added from outside. These are naturally processed and presented by the T cells and subsequently sequenced using 10X single-cell RNAseq and 10X single-cell TCRseq experiments. This method was successfully used to identify T cell functional states following antigen-stimulation in an ex vivo T-cell activation system. We successfully identified functional gene clusters and molecular networks that are unique to CD8+ T cell exhausted state. The combined expression of our T cell exhaustion gene signature correlated with poor prognosis when applied to TCGA data of almost 1400 tumors from many different cancers. Furthermore, the molecular pathways identified in this study provide opportunities to develop novel therapeutic interventions specially targeting dysfunctional T cells in cancers thereby enhancing the efficacy of checkpoint inhibitors.6
References
- 1. Davda, J. et al. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J. Immunother. Cancer 7, 105 (2019).
- 2. OncoPeptVAC: A robust TCR binding algorithm to prioritize neoepitope using tumor mutation (DNAseq) and gene expression (RNAseq) data. 3 1, 223–223 (2017).
- 3. Majumder, S. et al. A cancer vaccine approach for personalized treatment of Lynch Syndrome. Sci. Rep. 8, 12122 (2018).
- 4. Mahajan, S. et al. Immunodominant T-cell epitopes from the SARS-CoV-2 spike antigen reveal robust pre-existing T-cell immunity in unexposed individuals. Sci. Rep. 11, 13164 (2021).
- 5. Bhojak, K. et al. Immunodominant influenza epitope GILGFVFTL engage common and divergent TCRs when presented as a 9-mer or a 15-mer peptide. http://biorxiv.org/lookup/doi/10.1101/2022.07.11.499638 (2022) doi:10.1101/2022.07.11.499638.
- 6. Shi, X. et al. Abstract 4943: Leveraging single-cell sequencing to discover novel exhaustion markers of CD8 T cells. Cancer Res. 79, 4943 (2019).
#T cells, #T-cell immunity, #personalized medicine, #precision therapies, #tumor mutation burden, #genomic medicine, #T-cell Activation, #anti-PD1, #anti-CTLA4, #RNA-seq data, #immune checkpoints, #genomic profiling