Single cell sequencing is a cutting-edge technique used in molecular biology that enables the sequencing of the transcriptome of individual cells. In traditional bulk sequencing techniques, RNA is extracted from a large group of cells, and then sequenced as a whole. However, single cell sequencing allows researchers to analyze the genetic material of individual cells, providing a much more detailed and precise understanding of the diversity and heterogeneity of cell populations.
By Derek Vargas, Scientific Affairs, MedGenome Inc
Single cell sequencing is a cutting-edge technique used in molecular biology that enables the sequencing of the transcriptome of individual cells. In traditional bulk sequencing techniques, RNA is extracted from a large group of cells, and then sequenced as a whole. However, single cell sequencing allows researchers to analyze the genetic material of individual cells, providing a much more detailed and precise understanding of the diversity and heterogeneity of cell populations.
Single cell sequencing involves several steps, including isolating individual cells from a sample, lysing the cells to release their genetic material, amplifying the RNA to generate enough material for sequencing, and then sequencing the material using high-throughput sequencing technologies. There are several different types of single cell sequencing, each with its own strengths and limitations.
Single cell sequencing has many potential applications in basic research and clinical settings. It can be used to study complex biological processes such as embryonic development, cancer progression, and immune system function. It can also be used in research settings to identify rare cell types or genetic mutations that may be missed using traditional sequencing methods. MedGenome offers end-to-end single cell sequencing services using the 10X Genomics platform. Our extensive experience with single cell work allows us to process many samples with fast turnaround time.
Single Cell Sequencing at MedGenome
Despite the many advantages of single cell sequencing, there are also several challenges associated with this technique. Some of these challenges include:
1. High cost: Single cell sequencing can be expensive, as it requires specialized equipment and reagents, as well as significant computational resources for data analysis.
2.Low throughput: Single cell sequencing is a time-consuming process that can only sequence a limited number of cells at a time, which can limit the statistical power of the analysis.
3. Difficulty processing fresh samples: Many research labs are not able to process tissue samples quickly while cells are still viable. This may impact overall data quality
4. Limited availability of fresh samples: Single cell sequencing generally requires high quality fresh/frozen samples. This is often not possible for scientists who focus their studies on retrospective research using biobanked samples.
As a leading provider of single cell sequencing services, MedGenome has developed solutions to many of these problems. Using a combination of skilled scientists and automation, we can support large scale projects while keeping costs low. We also have protocols in place to help researchers preserve their precious samples until they can ship them to our labs. This has allowed MedGenome to process hundreds of single cell samples every year.
10X Genomics Single Cell Flex Kit
As I mentioned, a major limitation of single cell sequencing is that it can be difficult for researchers to preserve samples for processing. There are also researchers doing retrospective studies and getting new samples is impossible. 10X Genomics has recently released a new Single Cell Flex kit which addresses these issues. With this new Flex kit, cells are fixed and permeabilized and can be safely stored or transported without compromising data quality. Once ready to proceed, samples are hybridized to probe sets and may be processed individually (singleplex workflow) or pooled with up to sixteen samples in a single lane of a Chromium chip (multiplex workflow). During GEM generation the probe sets are ligated and extended to incorporate unique barcodes. Libraries are then prepared, sequenced, and analyzed using 10x Genomics Cell Ranger and other bioinformatics tools.
The ability to fix cells and tissues will help scientists who aren’t able to immediately process their samples for single cell sequencing. It is even possible to stain the cells with Totalseq antibodies prior to fixation. The cells can then be stored for months at -80 oC. This will benefit researchers doing longitudinal studies where samples are expected to be collected over a long period of time. In this case the samples can be fixed, frozen, and then shipped once the entire experiment is completed.
The Single Cell Flex kit also allows profiling of FFPE tissues. This workflow uses the Miltenyi Gentlemacs Dissociator (available at MedGenome) to dissociate FFPE scrolls into single cell suspensions. Since the Single Cell Flex kit is probe-based, it allows for a pseudo-transcriptome profiling of these highly degraded samples and allows researchers to gain insights into the cell populations of tissues that may have been collected several years ago.
MedGenome has always been an early adopter of single cell sequencing technologies. We started offering single cell gene expression, and have expanded over the years to support multiomic analysis of samples- including CITE-seq, immune profiling, and ATAC-seq. We have also adopted technologies for tissue dissociation and nuclei isolation. Our goal is to make single cell sequencing technologies available and affordable to genomics research labs, and to support research projects regardless of sample limitations or experiment complexity. Our team is excited about the new Single Cell Flex kit and expect to be offering this service very soon.
Spatial transcriptomics is a technology that allows the analysis of gene expression patterns within a tissue sample in their spatial context. It enables researchers to obtain a comprehensive and high-resolution view of the transcriptome, the set of all expressed genes, across different regions of the tissue. In traditional transcriptomics, gene expression is measured from homogenized cell populations, which can mask important differences in gene expression between different cell types and regions. Spatial transcriptomics, on the other hand, allows researchers to analyze gene expression patterns in intact tissue sections while retaining their spatial information.
By Derek Vargas, Scientific Affairs, MedGenome Inc
Spatial transcriptomics is a technology that allows the analysis of gene expression patterns within a tissue sample in their spatial context. It enables researchers to obtain a comprehensive and high-resolution view of the transcriptome, the set of all expressed genes, across different regions of the tissue. In traditional transcriptomics, gene expression is measured from homogenized cell populations, which can mask important differences in gene expression between different cell types and regions. Spatial transcriptomics, on the other hand, allows researchers to analyze gene expression patterns in intact tissue sections while retaining their spatial information.
Spatial transcriptomics typically involves the following steps:
• Preparation of the tissue sample: Tissue sections are cut and placed on a surface that contains oligonucleotide-labeled spots or barcode arrays.
• Capture of mRNA molecules: The mRNA molecules in the tissue section are captured and attached to the labeled spots, allowing for the spatial location of the mRNA to be retained.
• High-throughput sequencing: The captured mRNA molecules are amplified and subjected to high-throughput sequencing, generating a large amount of data.
• Data analysis: The sequencing data is analyzed to identify the expression levels of different genes and their spatial distribution within the tissue section.
Overall, spatial transcriptomics provides a powerful tool for studying complex tissues, such as the brain, where multiple cell types with distinct gene expression profiles are tightly organized in intricate spatial arrangements. The technology can help researchers to gain new insights into the molecular mechanisms of development, disease, and tissue function. There are several platforms available for spatial transcriptomics, however the two most widely used platforms are Visium (10x Genomics) and GeoMx (Nanostring).
Visium Advances Biomarker Discovery
Visium is a spatial transcriptomics technology developed by 10x Genomics that enables high-throughput analysis of gene expression in intact tissue sections. Visium builds on the principle of spatial transcriptomics and allows for the analysis of the whole transcriptome in a spatially resolved manner, meaning that researchers can obtain detailed information on the gene expression patterns within a tissue sample while maintaining their spatial context.
The Visium platform is based on the capture of mRNA molecules on an array of polymeric spots on a glass slide. The captured mRNA molecules are then barcoded, reverse transcribed, and amplified. The amplified cDNA is sequenced using high-throughput sequencing technologies, generating millions of reads that are aligned to a reference genome. The resulting data can be visualized and analyzed using various software tools provided by 10x Genomics or other bioinformatics platforms. The technology can be used to study complex tissues and biological processes, such as development, disease, and tumor microenvironments, by providing information on the expression levels of thousands of genes in each region of the tissue section.
Nanostring GeoMx Allows Targeted Analysis of Spatial Transcriptome
NanoString’s GeoMx is another spatial profiling technology that enables the analysis of gene expression at high resolution within a tissue sample while retaining its spatial context. The technology is based on the digital barcoding and imaging of RNA molecules in situ, allowing for the precise spatial localization of gene expression patterns within a tissue sample. The GeoMx system utilizes a set of molecular probes that are pre-designed or customized for specific gene targets. The probes are attached to a surface and hybridized to the RNA molecules in the tissue sample, creating a unique barcode sequence for each molecule. The barcoded RNA molecules are then imaged using a high-resolution imaging system, enabling the precise spatial localization of gene expression patterns in the tissue.
The GeoMx technology can be used to analyze hundreds to thousands of genes simultaneously, allowing researchers to gain a comprehensive view of the transcriptome across different regions of the tissue. The technology can be used to study various biological questions, such as the identification of cell types, the characterization of disease-associated gene expression patterns, and the discovery of new biomarkers for diagnosis and therapy. The GeoMx system is also compatible with other NanoString technologies, such as the nCounter platform, enabling researchers to combine spatial profiling with digital quantification of gene expression in the same sample. The technology has applications in various fields, including oncology, immunology, and neuroscience, and can be used in both research and clinical settings.
Conclusion
Visium and GeoMx are both spatial profiling technologies, but there are some key differences between the two platforms. Visium doesn’t require any large lab equipment. The chemistry takes place on a special microscope slide and the workflow can be completed with standard lab equipment found in many cell biology labs. On the other hand, GeoMx does require special machinery, which can make it more difficult for smaller labs to utilize this technology. Another major difference is that Visium takes an unbiased approach to spatially profiling tissues; any tissue placed in the capture area of the slide will be sequenced. GeoMx requires some prior knowledge of the tissue since regions of interest must be chosen. This difference makes Visium a great tool for discovery research, while GeoMx is great for clinical research.
Overall, both Visium and GeoMx are powerful tools for studying gene expression patterns and cellular heterogeneity within complex tissues. The choice of platform depends on the research question and the specific needs of the experiment, as each platform has its own strengths and limitations. Currently, MedGenome is offering full bioinformatics services related to Visium datasets. There are many standard and custom analysis options available to accommodate most projects. Additionally, our genomics lab is in the process of adopting spatial transcriptomics technologies. We expect end-to-end Visium spatial profiling services to be offered in the near future.
Next-generation sequencing (NGS) data is being increasingly used in clinical diagnosis to identify genetic variation that can be a cause for the disease. A major challenge in using NGS data in a clinical setting is to make the right interpretation because of its huge size and complexity. Also, there are possibilities of technical errors during the sample processing and/or sequencing stage that may be inherent to the kind of sequencing technology used. Therefore, the use of reference standards is of paramount importance to mitigate and minimize these errors.
By Archana Deshpande, QA Manager, MedGenome Inc
Introduction
Next-generation sequencing (NGS) data is being increasingly used in clinical diagnosis to identify genetic variation that can be a cause for the disease. A major challenge in using NGS data in a clinical setting is to make the right interpretation because of its huge size and complexity. Also, there are possibilities of technical errors during the sample processing and/or sequencing stage that may be inherent to the kind of sequencing technology used. Therefore, the use of reference standards is of paramount importance to mitigate and minimize these errors.
Reference standards play an important role in the life cycle of a typical NGS method implementation before clinical application. A typical NGS assay life cycle includes assay development, optimization, validation, and continuous quality management – standard is of consequence in all these aspects ranging from assay validation to technical validation to sample processing. This article will discuss the general selection of these reference standards and describe in detail the results of technical validation of a target-capture assay (Illumina’s TruSight Oncology 500 panel) that was performed at MedGenome Labs.
NIST and GIAB Standards
There are several consortiums like GIAB (Genome in a Bottle) and companies (Horizon Diagnostics and SeraCare) that have developed DNA reference material over the years to support clinical translation of whole genome sequencing. NIST (National Institute for Standards and Technology) also had a program to develop whole human genome reference materials. In general, reference standards are well-characterized samples, that are consistent and stable over time. Essentially, the DNA reference is characterized by collating data from various sequencing and bioinformatics methods and from multiple datasets to yield highly confident genotype calls. This data can then be used by laboratories for evaluating assay performance and accreditation agencies for benchmarking results.
In addition to being homogeneous and stable, NIST has defined standards with values that they have certified indicating confidence in their accuracy. This certification indicates that NIST has fully investigated and accounted for all known or suspected sources of bias seen in the data.
MedGenome Validation Data
At MedGenome Labs, we have validated Illumina’s TSO 500 workflow and pipeline using reference samples from SeraCare. TSO 500 is a target-capture based panel that interrogates multiple biomarkers and tumor types; it identifies all relevant DNA and RNA variants implicated in various solid tumor types. Thus, it allows for in-house comprehensive genomic profiling of tumor samples. It also accurately measures key current immuno-oncology biomarkers: microsatellite instability (MSI) and tumor mutational burden (TMB). The other advantage is that the assay has a ctDNA panel that can be used for liquid biopsies.
The workflow for either TSO 500 is a hybrid capture protocol, and we validated our process by using three control ctDNA (with different allele frequencies 0.1%, 0.5% and Wild Type) from SeraCare. The data generated was from as little as 30 ng of starting input.
We performed analysis of the Somatic mutations with gene list present in Seraseq ctDNA Complete Mutation Mix AF 0.5%, AF 0.1% and WT. The analysis included sensitivity, specificity, positive predictive value, and inter-run comparison. Below are some of the results that we obtained.
Library Quality Report
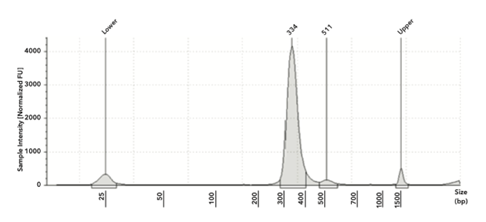
The libraries created from SeraSeq controls ranged from 23 to 46 nM and had an average size of 330 bp with ~200 bp insert size (see Figure 1 for example). This library size fell in the range that is specified in the TSO 500 protocol.
Figure 1: Example of a final library from SeraSeq ctDNA control (this was 0.5% allelle frequency with a peak at 334 bp)
The reference SeraSeq sets were analyzed using TruSight Oncology 500 ctDNA local app. The results were compared with the reference set data provided by the vendor. We were able to obtain 100% sensitivity and specificity for all 3 controls of dataset. The variants are also represented in the lollipop plot which is given below along with the sensitivity and specificity information for the controls (Table 1).
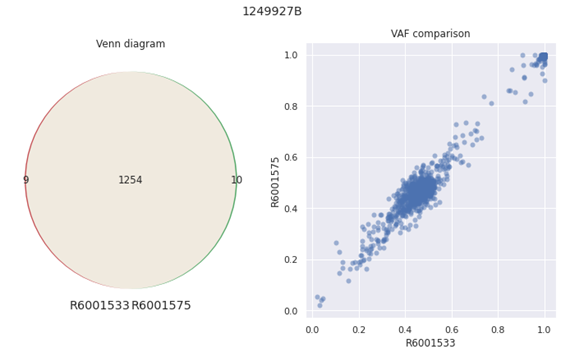
For the inter-run comparison, the samples were compared for the tumor mutation burden count and variant allele frequency (VAF) of the same sample between two runs. The Identified variants were found to be nearly identical and VAF values are highly correlated (See Fig 2 below).
Figure 2: VAF comparison illustration for inter-run comparison
Our process workflow for Illumina’s TSO 500 panel passed the technical validation and we have started offering the TSO500 panel for both solid tumor and ctDNA as part of our services for clients.
Conclusion
DNA reference standards are vital for translational medicine as well as research and MedGenome uses commercially available reference standards to perform technical validation wherever they are available. The validation allows us to have confidence in our process workflows and generated data.
References
1. Genomic Reference Materials for Clinical Application Justin Zook and Marc Salit Biosystems and Biomaterials Division, National Institute of Standards and Technology, 100 Bureau Dr., Gaithersburg, MD 20899
2. Reference standards for next-generation sequencing Simon A. Hardwick, Ira W. Deveson and Tim R. Mercer, Nature Reviews Genetics · June 2017
NGS technologies is at the forefront of Biological Research. They produce enormous data running into gigabases in a single round of sequencing. However, several sequencing artifacts such as read errors (base calling errors and small insertions/deletions), poor quality reads and primer/adaptor contamination are quite common with the NGS data obtained after sequencing.
By Parimala Nagaraja, Scientist, NGS, MedGenome Inc.
NGS technologies is at the forefront of Biological Research. They produce enormous data running into gigabases in a single round of sequencing. However, several sequencing artifacts such as read errors (base calling errors and small insertions/deletions), poor quality reads and primer/adaptor contamination are quite common with the NGS data obtained after sequencing. It can impose significant impact on the downstream analysis such as sequence assembly, single nucleotide polymorphisms (SNP) identification and gene expression studies.
Quality control metrics play a critical role in ensuring to minimise the number of errors and help in achieving high quality data for a successful experimental study. MedGenome strives to maintain strict guidelines in terms of QC metrics to achieve high quality data for our clientele.
QC metrics are mainly applied at 3 levels:
• Sample QC (DNA/RNA)
• Library QC
• Sequencing QC
Sample QC
An Ideal NGS assay would require high quality DNA/RNA which is usually determined using Tapestation/Bioanalyzer that provide the DIN/RIN (DNA/RNA Integrity number) values ranging between 1-10, where 10 is the highest quality sample and 1 is the highly degraded and poor-quality samples.
Depending on the assay type and Sample source, MedGenome has a set of guidelines in terms of Quantity, Quality and Volumes for the clients. At MedGenome, all samples are first subjected to QC using Qubit to determine the quantity and Tapestation/Bioanalyzer to determine the quality.
Based on the QC determined, samples are classified as a Pass or Marginal or Fail. Replacement samples are usually requested for the samples that failed Sample QC. For Marginal samples, replacements are highly encouraged, else they will be proceeded to library preparation after client’s approval.
Library QC
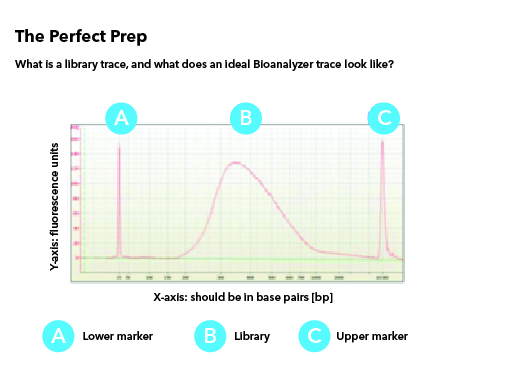
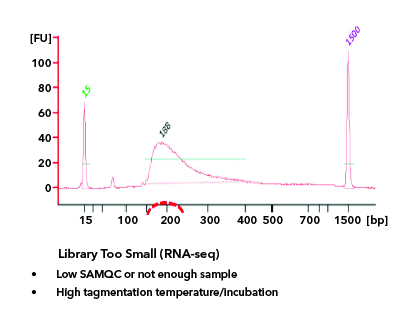
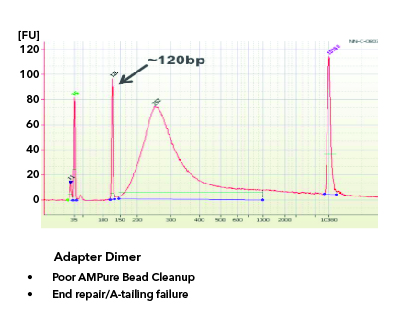
All libraries which are prepared in-house are checked for their quality using Tapestation/Bioanalyzer and quantified using Qubit. Tapestation and Bioanalyzer results are thoroughly reviewed for the expected library size, adapter contamination, primer dimers and PCR artifacts before they are pooled and loaded onto the sequencer. MedGenome also offers sequencing support for Premade libraries which are prepared by various clientele based on their project requirements. All premade libraries are also subjected to MedGenome QC methodologies and are diligently reviewed and classified as Pass or Marginal or Fail before sequencing. Following Images provides an example for Good vs Bad Library QC.
Figure 1: Example of a Good library QC. Library is in the expected size, in a single bell curve and devoid of Adapter dimer contamination.Figure 2: Example 1 for Bad Library QCFigure 3: Example 2 for Bad Library QCFigure 4: Example 3 for Bad Library QC
Sequencing QC
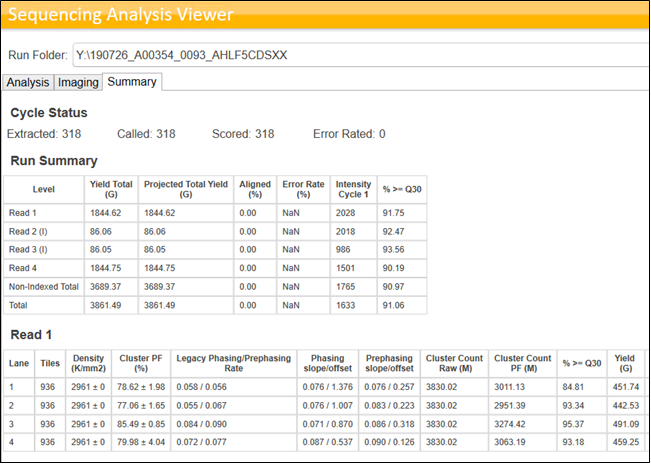
Illumina facilitates the users to monitor the runs in real time without interfering with the run performance using a software called Sequencing Analysis Viewer (SAV). This software is compatible with all HiSeq, NextSeq, MiSeq and NovaSeq platforms. The following table describes the features used for evaluating the Sequencing QC:
Table 1: Different terms and their corresponding definitions as viewed in SAV.
Term
Definition
Intensity
The 90% percentile extracted intensity for a given image (lane/tile/cycle/channel combination). On platforms using four-channel sequencing, 4 channels (A, C, G, and T) are shown.
FWHM
The average full width of clusters at half maximum (representing their approximate size in pixels).
% Base
The percentage of clusters for which the selected base has been called.
%Q >/= 20, %Q >/=30
The percentage of bases with a Phred or Q quality score of 20 or 30 or higher, respectively
Density
The density of clusters for each tile (in thousands per mm2).
Density PF
The density of clusters passing filter for each tile (in thousands per mm2).
Clusters
The number of clusters for each tile (in millions).
Clusters PF
The number of clusters passing filter for each tile (in millions. (Metrics given in below images)
% Pass Filter
The percentage of clusters passing the Chastity filter (Metrics given in below images)
% Phasing, % Prephasing
The average rate (percentage per cycle) at which molecules in a cluster fall behind (phasing) or jump ahead (prephasing) during the run.
% Aligned
The percentage of the passing filter clusters that aligned to the PhiX genome.
Error rate
The calculated error rate, as determined by the PhiX alignment. Subsequent columns display the error rate for cycles 1–35, 1–75, and 1–100.
Yield Total
The number of bases sequenced, which is updated as the run Progresses. (Metrics given in below images)
Projected Total Yield
The projected number of bases expected to be sequenced at the end of the run.
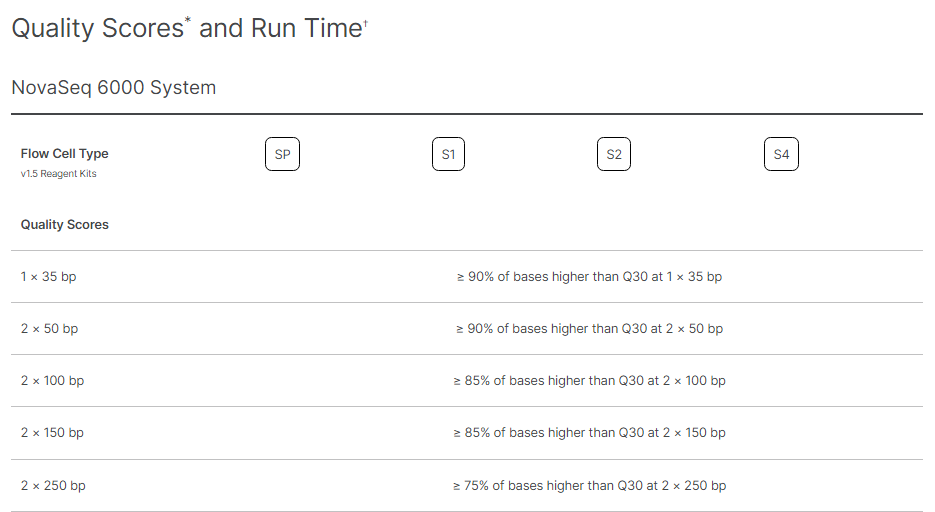
Illumina provides the standardised expectations of reads outputs, Reads Passing Filters, and Quality Scores for each Flow Cell type on every sequencing platform. Following Images provide the metrics for different flowcells on NovaSeq 6000.
Figure 5: NovaSeq 6000 read Read output Specifications Image Source: https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.htmlFigure 6: Total Reads that passes the filter on NovaSeq Platform Image Source: https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.htmlFigure 7: Illumina Standard for Read Quality on NovaSeq 6000 Image Source: https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.html
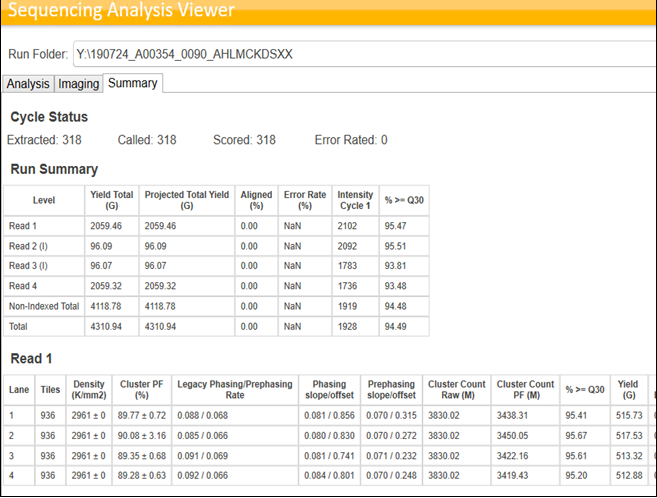
Sequencing QC also depends on the library types pooled into the same lane or Flow Cell. If libraries prepared using the same protocol (For ex: Illumina Stranded mRNA) are pooled and sequenced, we can see NovaSeq outperforming the Illumina specifications. However, this is usually not the case in an ideal world for any NGS service providing company with high throughput fast paced Turn Around Times. Hence, when multiple libraries of different library types are pooled, it is expected to see the variations in the run performances and the data yields. Following images provide an example of the Sequencing stats achieved by pooling similar libraries and Mixed libraries.
Figure 8: SAV Stats for the run having same library type. Example for the best stats viewed on SAV for an S4 run performed at MedGenome. Cluster PF(%)<85%, Total yield per lane (~3.4 Billion PE) exceeding the Illumina specifications, %>=30 above 95%.Figure 9: SAV stats for the sequencing run having mixed library types. Cluster PF(%) ~80%, Total yield per lane (~3 Billion PE) %>=30 ranging from 85% to 93%. However, this run can be classified as “Good run” as it has met all the Illumina Standard metrics.
Quality Control of the Sequencing raw data
Raw data quality control should be the initial step of data analysis for any successful study. There are several tools that are publicly available for conducting quality control on raw FASTQ files. FastQC developed by Babraham Institute bioinformatics group is one of the most popular tools that offers QC control parameters such as average base quality score per read, the GC content distribution and identification of the most duplicated reads.
The important parameters to check for raw sequencing data quality are:
• Base Quality
• Nucleotide distribution
• %GC distribution
• PCR duplicates
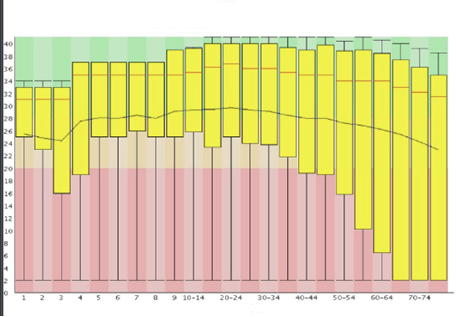
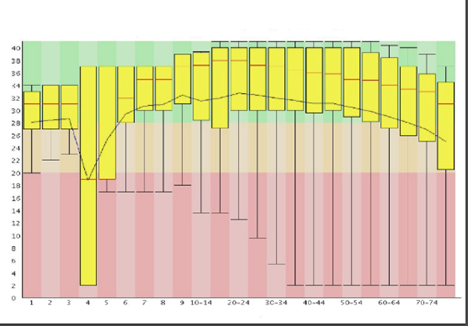
Base Quality check:
A common way to visualize base quality is to draw a base Q-score versus cycle plot. Sequencing data generated on Illumina platforms tend to observe a median base quality score between 35 and 40 in the Phred scale. Large variations in base quality scores (Figure 10a) usually indicate poor Library QC. Sudden drop in the Quality scores (Figure 10b) usually indicate Adapter dimer contaminations or Fluidics issue in the instrument. For paired-end reads, it is common to observe higher quality in the first end of the read than the second end owing to the amount of time the template was on the instrument and increasing laser exposure over time.
Figure 10a: Quality score variations due to poor Library QC Image Source: Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2014 Nov;15(6):879-89. doi: 10.1093/bib/bbt069. Epub 2013 Sep 24. PMID: 24067931; PMCID: PMC4492405.Figure 10b: Quality drop due to Adapter dimer contamination. Image Source: Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2014 Nov;15(6):879-89. doi: 10.1093/bib/bbt069. Epub 2013 Sep 24. PMID: 24067931; PMCID: PMC4492405.
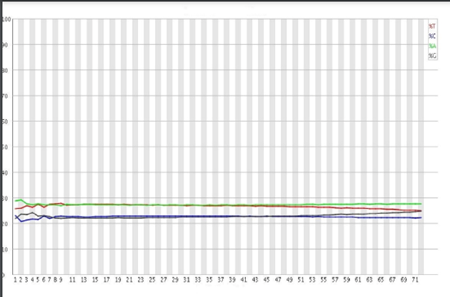
Nucleotide Distribution
This parameter is useful for Whole genome and Whole exome libraries (High diversity) but not for Amplicons or RNA libraries (Medium-Low diversity). For a perfect sequencing run, the distribution of the four nucleotides (A T C G) across all reads should remain relatively stable (Figure 11)
Figure 11: Nucleotide distribution for a perfect sequencing run Image Source: Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2014 Nov;15(6):879-89. doi: 10.1093/bib/bbt069. Epub 2013 Sep 24. PMID: 24067931; PMCID: PMC4492405.
%GC Distribution
The percentage of GC in the genome varies across species and across the regions of each genome. For exome regions, the GC content is about 49–51%, while for whole-genome sequencing (Human), the GC content is around 38–40%. Abnormal GC content percentage (>10% deviation from normal range), can indicate contamination.
PCR Duplicates
PCR duplicates arise during library preparation when PCR amplifies the fragments with adapters. Presence of PCR duplicates can lead to potential biases in variant calling algorithms. Hence these are removed by most of the Bioinformatic analysis pipelines during the pre-processing of the data. General causes for high rate of PCR duplicates are Low input quantity, Over sequencing, too many PCR cycles, Low pre-enrichment yield/final library yield, and short library fragments.
Conclusion
MedGenome strives to follow all the best practices in Lab and QC methodologies. Apart from just performing QC, we also interpret and communicate with the client regarding any deviations from MedGenome’s QC standards and recommend the best possible actions to proceed. After the sequencing is performed to the best of our abilities, the raw data is thoroughly reviewed as per Illumina’s standards prior to the data being shared with clients. MedGenome also offers data and sample storage facilities as per clients’ requests.
5. Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2014 Nov;15(6):879-89. doi: 10.1093/bib/bbt069. Epub 2013 Sep 24. PMID: 24067931; PMCID: PMC4492405.
Our journey in 2022 was focused on providing the utmost customer experience for the services and solutions that we delivered to you. Along with expanding our portfolio of services and solutions – the tissue dissociation and nuclei isolation services to support our single cell customers, streamlined antibody discovery using high-throughput single B cell receptor sequencing, TSO500 targeted panels for oncology research, single cell and bulk epigenetics assays.
By Hiranjith GH, VP & Head of Research Services, MedGenome Inc.
2022 was an eventful year,
Our journey in 2022 was focused on providing the utmost customer experience for the services and solutions that we delivered to you. Along with expanding our portfolio of services and solutions – the tissue dissociation and nuclei isolation services to support our single cell customers, streamlined antibody discovery using high-throughput single B cell receptor sequencing, TSO500 targeted panels for oncology research, single cell and bulk epigenetics assays. We improved our turn-around times on bulk transcriptomics, whole exome and whole genome projects by incorporating automation at multiple project stages and installing new sequencing capacity in 2022. Our sequencing team has delivered high-quality data consistently for a variety of library types throughout the year.
We also rolled out a series of advanced and interactive analyses reports for each of our assays through our unique ManGo platform with novel data representations. This scale up on our bioinformatics capabilities is in line with our objective to support biologists and researchers to maximize the utility from genomic data.
We partnered with our customers to discuss the data and the subsequent analyses after each project is delivered to ensure that the results meet the researcher needs.
We built streamlined systems and communication processes for sample and data management with our customers, which allowed us to build transparency and a trusted relationship.
The MedGenome Research Team
In 2023,
We expect to continue to offer high quality support to your projects in 2023. We will be spending lab resources to optimize spatial transcriptomics and Hi-C assays in-house in 2023 to expand our services portfolio. With supporting bioinformatics analyses and tools to provide end-to-end service to our customers.
We will engage with customers on antibody discovery solutions and protein expression services given the highly experienced R&D talent that we have at MedGenome.
With growing sample volumes, we are committed to investing in our sequencing capacity even further in 2023 – to help us maintain the turn-around times (TAT) for the projects. We will discontinue our HiSeq X service by the end of this year.
We are also a preferred partner to customers who are looking to access South Asian genomic datasets in specific diseases areas (rare diseases, oncology, neuro-degenerative disorders, blood disorders, metabolic diseases) for discovery or genetic modifier studies. With a vast network of hospital collaborations in India, MedGenome is able to accelerate these studies with high impact.
Pandemic has tested our systems, processes and quality of team members beyond doubt. It has shown the importance of value added engagements with our customers that MedGenome strives for. Going into 2023, MedGenome is looking forward to continuing those relationships to advance genomics research by our customers.
The discovery of genetic and epigenetic mechanisms underlying the onset and progression of numerous diseases, including cancer, has helped redefine clinical research, diagnostic and treatment paradigms. Oncology research and diagnostics have undergone radical changes because of the development of next-generation sequencing (NGS). NGS has improved rationally designed personalized cancer medicine by identifying novel cancer mutations, detecting circulating tumor DNA (ctDNA), and discovering causative mutations for hereditary cancer syndrome. With NGS, it is now possible to sequence the whole genome, whole exome, whole transcriptome, or just targeted genes to provide detailed genomic landscape descriptions for many cancers.
By Dr. Chaitanya Ekkirala, Lab Director, NGS Operations, MedGenome Inc
Overview
The discovery of genetic and epigenetic mechanisms underlying the onset and progression of numerous diseases, including cancer, has helped redefine clinical research, diagnostic and treatment paradigms. Oncology research and diagnostics have undergone radical changes because of the development of next-generation sequencing (NGS). NGS has improved rationally designed personalized cancer medicine by identifying novel cancer mutations, detecting circulating tumor DNA (ctDNA), and discovering causative mutations for hereditary cancer syndrome. With NGS, it is now possible to sequence the whole genome, whole exome, whole transcriptome, or just targeted genes to provide detailed genomic landscape descriptions for many cancers.
MedGenome is committed to providing the highest-quality NGS services for research and clinical development. Our expert scientific team and laboratory facility with cutting-edge sequencing platforms, including NovaSeq, MiSeq, and 10X Chromium Controller, guarantee highly optimized protocols, customized solutions, and quick turnaround times.
Targeted panel for Oncology mutational profiling using TSO-500
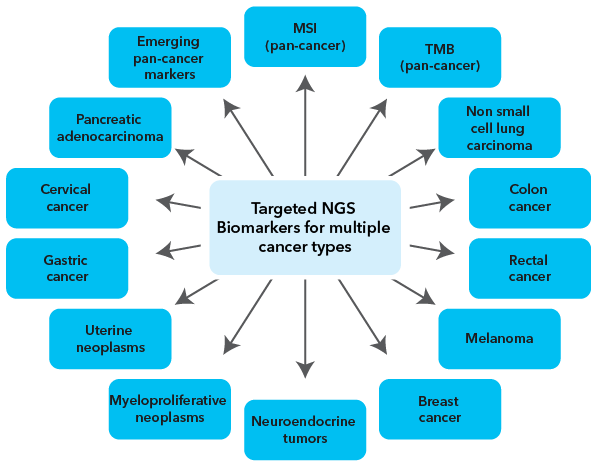
MedGenome offers comprehensive genomic profiling of tumor samples using the TruSight Oncology 500 (TSO-500) from Illumina. The TSO-500 is a pan-cancer NGS assay with broad availability, rapid turnaround time, and a standardized bioinformatics pipeline that identify key genomic signatures for clinical research and immuno-oncology. The panel includes 523 cancer-relevant gene variants and 55 RNA variants that provide comprehensive coverage of biomarkers (Figure 1) frequently mutated in multiple cancer types.
Figure 1: : The tumor genomic profiling using TSO-500 assay includes pan-cancer markers and gene variants validated in 11 solid tumor types. The marker selection is based on the current guidelines and emerging biomarkers detailed by the National Comprehensive Cancer Network (NCCN) guidelines and reports from over 1600 clinical trials.
TSO-500 employs a highly standardized single integrated workflow for both DNA and RNA input material. Library preparation involves a hybridization capture-based target enrichment strategy. High analytical specificity is achieved by adding unique molecular identifiers (UMIs) during library prep, which allows the detection of gene variants even at low variant allele frequency (VAF) while simultaneously suppressing errors. Sequencing reactions are carried out using fluorescence-labelled oligonucleotides, and an off-the-shelf bioinformatics pipeline provides robust and reliable results.
Features
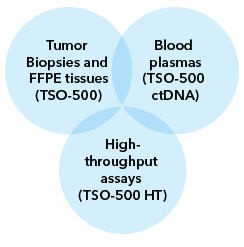
• Variable sample types and throughput MedGenome offers flexible and scalable genomic profiling from tumor biopsies, FFPE tissues, and liquid biopsies (to detect ctDNA) using the Illumina TruSight Oncology 500 portfolio (Figure 2). The platform also allows analysis of sample sizes ranging from 8-192 samples, even allowing accurate detection in low input samples (from 30ng DNA and 40ng RNA input material).
Figure 1: The same oncology panel can be efficiently installed at scale for various sample types to meet your research requirements
• Easy ctDNA detection from liquid biopsies Noninvasive plasma-based assays have emerged as an important complementary diagnostic approach to tissue-based assays which is not feasible for repeated sampling or inaccessible tissues. Further, single biopsies lack information on tumor heterogeneity. Blood plasma contains tumor cell fragments and DNA from apoptotic or necrotic cancer cells providing information on cancer aggressiveness, progression and therapeutic outcomes. The TSO-500 ctDNA assay enables non-invasive, comprehensive genomic profiling of ctDNA from simple blood draws to evaluate >500 gene variant classes in a single assay.
• Optimized data Analysis The variant calling algorithms are optimized to eliminate errors, artefacts, and germline variants for high accuracy and analytical specificity (99.9998%). Data interpretation and reporting are powered by PierianDx Clinical Genomics Workspace (CGW), which filters and prioritizes biologically relevant variants providing an automated and customizable genomic report.
• Accurate TMB and MSI analysis TSO-500 assay implements an error-corrected sequencing and informatic pipeline that provides an accurate quantitative score for MSI status and a precise and reproducible TMB value. TMB calculation involves the measurement of both nonsynonymous and synonymous SNVs and InDels based on specific criteria. The results were shown to have high concordance with whole-exome studies.
• High sensitivity Using the TSO-500 platform, we provide highly sensitive variant detection for CNVs with a limit of detection at 2.2× fold-change. In addition, the TSO-500 library prep protocol implements high binding specificity to hybridize to targets containing small mutations and SNVs, even from low-quality DNA samples and FFPE tissues. The assay reproducibility has been verified in FFPE samples with a VAF as low as 5%. Furthermore, with regards to the detection of RNA fusions, the hybrid-capture method accurately captures gene fusions from both known and novel fusion gene partners, even from FFPE samples where RNA yields can be >= 40ng.
The MedGenome Advantage
MedGenome offers end-to-end customized solutions for comprehensive genomic profiling of small and large-scale tumor samples to accelerate your clinical research and diagnostics R&D. We have expertise in processing a variety of sample types, including biopsies, FFPE tissue (DNA & RNA), and blood plasma (ctDNA) for DNA sequencing. Our scientific team can also provide solutions to challenging sample processing and high-throughput samples.
Utilizing the TruSight targeted panel we provide exclusive services and advantages:
• Multiplexing solutions – Save time and samples by analyzing multiple tumor variant types in 523 genes in a single assay
• Speed – Streamlined validated workflow with quick turnaround times for TSO-500 assay
• Powerful bioinformatics pipelines – Additional insights on drug-gene interactions, identification of actionable mutations, and comprehensive reports
• High-throughput processing – Scientific expertise and state-of-the-art lab facility for large sample sizes suitable for clinical research
• TST170 Panel for ctDNA – We have validated the TruSight Tumor 170 panel on ctDNA samples for assessment of SNVs and indels in 151 genes, amplifications in 59 genes, and fusions plus splice variants in 55 genes.
Need more insights on tumor profiling using NGS?
Click here to get in touch with our expert scientific team for unique solutions to your research. You can also email us at research@medgenome.com for any queries and further details.
4. Wei, B., Kang, J., Kibukawa, M., Arreaza, G., Maguire, M., Chen, L., Qiu, P., Lang, L., Aurora-Garg, D., Cristescu, R., & Levitan, D. (2022). Evaluation of the TruSight Oncology 500 Assay for Routine Clinical Testing of Tumor Mutational Burden and Clinical Utility for Predicting Response to Pembrolizumab. The Journal of molecular diagnostics: JMD, 24(6), 600–608. https://doi.org/10.1016/j.jmoldx.2022.01.008
Alzheimer’s disease (AD) has long been one of the great challenges in medicine and imposes a constant burden on our aging population. Recent statistics show that approximately 50 million people worldwide suffer from AD or some other form of dementia. The World Health Organization has estimated that the total number of people with dementia worldwide will reach 82 million by 2030 and 152 million by 2050. Of the top 10 leading causes of death based on United States cancer statistics, cardiovascular disease ranks first, tumors rank second and AD ranks sixth.
By Dr. Anantha Kethireddy Ph. D., MedGenome Scientific Affairs
Why is Alzheimer’s relevant?
Alzheimer’s disease (AD) has long been one of the great challenges in medicine and imposes a constant burden on our aging population. Recent statistics show that approximately 50 million people worldwide suffer from AD or some other form of dementia. The World Health Organization has estimated that the total number of people with dementia worldwide will reach 82 million by 2030 and 152 million by 2050. Of the top 10 leading causes of death based on United States cancer statistics, cardiovascular disease ranks first, tumors rank second and AD ranks sixth.
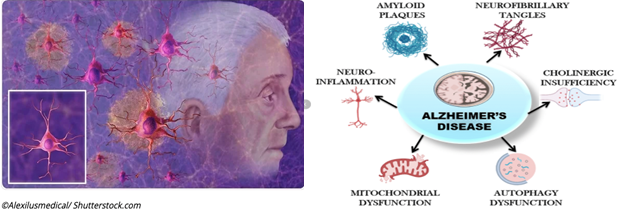
AD is a slowly progressing and eventually fatal neurodegenerative disorder and a major contributor of dementia leading to degeneration of neurons and their connections in parts of the brain involved in memory. The symptoms are impairment in thinking, remembering, reasoning, cognitive functions and behavior are known as dementia. Other diseases and conditions can also cause dementia, with AD being the most common cause of dementia in older adults. AD is not a normal part of aging. It’s the result of complex changes in the brain that starts years before symptoms appear and lead to loss of brain cells and connections. The hallmark of AD is the presence of plaques of the amyloid and neurofibrillary tangles of the phosphorylated protein tau. Much evidence suggests the involvement of neuroinflammation, multiple systemic comorbidities in the pathology of AD.
Single-cell sequencing technologies are proving to be a valuable tool in understanding the underlying molecular mechanisms of Alzheimer’s disease. By examining gene expression at the level of individual cells, single-cell sequencing allows for a more detailed and accurate analysis of how the disease progresses at a cellular level.
Since AD was first described in the early 1900s, clinicians and scientists all over the world dedicated their career to study the pathophysiology of this most common form of disease in the hope of developing methods of prevention, treatments to halt the progression and ultimately a cure. AD’s pathophysiology involves neuron-glia interactions, supported by transcriptomic and epigenomic analyses that reveal downregulation of neuronal functions and upregulation of innate immune responses in AD brains. Currently, there are some FDA approved tools that, when applicable, can be used to aid in diagnosis of AD symptoms (brain imaging), while other emerging biomarkers are promising but still under investigation (blood tests, genetic risk profiling).
Drugs do exist, if administered early enough they may help to treat the symptoms of early-stage AD and improve the person’s quality of life. Very recently, two pharmacological companies announced encouraging results from a clinical trial for patients with AD. A monoclonal antibody treatment, called lecanemab, suppressed cognitive decline by 27% in people with early-stage disease compared with those on a placebo after a year and half. Black or Hispanic populations also have a higher risk of Alzheimer’s disease than non-Hispanic white people, researchers don’t fully understand the reasons. There is no drug that works in everybody to stop or reverse AD.
Understanding AD
The molecular and cellular mechanisms of AD is incompletely understood. Typically, when scientists studied gene expression in the brain, they just mashed up the tissue and took average measurements from that mixture. Such “bulk” measurements are hard to interpret, and we lose the gene expression signals that come from individual cell types especially for lowly-represented cell types. Although numerous studies using bulk RNA-seq analysis have revealed dysfunctions of neurons and/or innate immune responses, they are unable to entangle the heterogeneity of different disease subtypes and distinct responses across cell types. Characterization of the heterogeneity of a region of the brain important for learning and memory, the first region affected in Alzheimer’s disease.
Single Cell Genomics
Even within a single brain region, there is a significant variation between the morphology, connectivity and electrophysical properties of individual neurons. A key step towards understanding the basic components of the nervous system is systematic classification of individual neurons. For cells to be classified on a molecular basis, gene expression must be assessed at single-cell resolution. Today’s high throughput technologies such as single cell and spatial multiomics are revolutionizing neurological research at single cell level resolution.
A cohesive demonstration of how gene expression is regulated within discrete cell types and specific anatomical regions of the brain during the early stages of AD is crucial to study the cellular heterogeneity of the brain by profiling tens of thousands of individual cells, capturing the molecular and cellular basis of AD and identifying novel therapeutic targets.
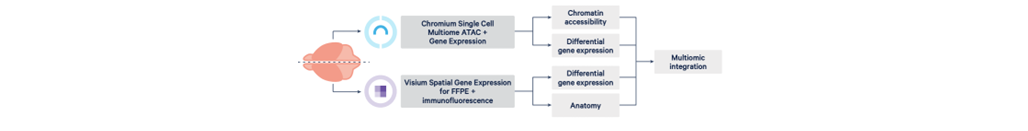
Figure 2: Multiomic integration from single brain (Image source: 10x Genomics)
Summary
Capturing a holistic view of cell type specific contributions to pathogenesis, mapping anatomical protein accumulation in the brain during disease progression, and understanding the relationship between abnormal protein accumulation and cellular phenotypes diagnosis at an early stage is very crucial to save the people from this dreadful disease.
By using Chromium Single Cell Multiome ATAC (Assay for Transposase-Accessible Chromatin) + Gene Expression (the multiome assay), which profiles open chromatin and gene expression from the same cell, and Visium Spatial Gene Expression for FFPE (Visium for FFPE) plus immunofluorescence (IF), which combines whole transcriptome spatial analysis with immunofluorescence protein detection will help to extract the shared and unique neurological disorders among different conditions.
The multidimensional datasets obtained through single cell genomics approaches will have major impact on biological research and clinical pathology. implementation and expansion of single-cell sequencing technologies will lead to vast improvements in the diagnosis and treatment of patients worldwide.
Modern medicine now derives its insights through the deeper understanding of the cellular and molecular mechanisms, which involves modification of the cellular behavior through targeted molecular approaches. Experimental biologists and clinicians now employ various molecular techniques to assess the intrinsic behavior of cells in a variety of ways, such as through analyses of genomic DNA sequences, chromatin structure, messenger RNA (mRNA) sequences, non-protein-coding RNA, protein expression, protein modifications and metabolites.
By Dr. Anantha Kethireddy Ph. D., MedGenome Scientific Affairs
Modern medicine now derives its insights through the deeper understanding of the cellular and molecular mechanisms, which involves modification of the cellular behavior through targeted molecular approaches. Experimental biologists and clinicians now employ various molecular techniques to assess the intrinsic behavior of cells in a variety of ways, such as through analyses of genomic DNA sequences, chromatin structure, messenger RNA (mRNA) sequences, non-protein-coding RNA, protein expression, protein modifications and metabolites.
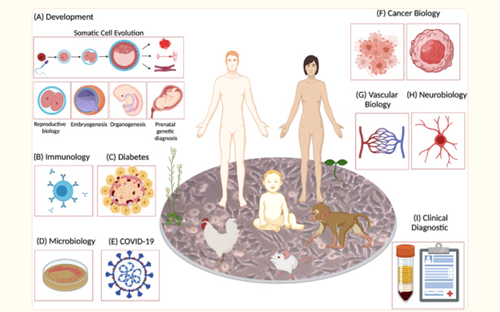
Today, single‐cell RNA expression profiling is rapidly becoming an irreplaceable method for various research including humans, animals and plants enabling more accurate, rapid identification of rare and novel cells in tissues like never before (Figure 1). Moreover, with this information about gene expression at mRNA and protein levels, metabolites, cell‐cell communication, and spatial landscape, it becomes possible to solve the puzzle of cell composition and functions in health and disease.
Although single cell sequencing studies have been conducted mostly by research groups over the past few years, it has become clear that biomedical researchers and clinicians can make important new discoveries using this powerful approach. While great promises have been demonstrated with the technological advancement in all areas, and its great potentials in transforming current protocols in diagnosis of the genetic drivers of the disease and treatment response mechanism from single cells to tissues.
Figure 1: Applications of Single–Cell sequencing Technology. (Source of the figure: Dragomirka Jovic et.al, Clin Transl Med, 2022 Mar,12)
Currently, there is a growing demand for single-cell technology, with nearly 200 different methods to profile not only transcriptomic but genetic, epigenetic, and proteomic information in individual cells.
An Overview of Single–Cell Technologies:
Single–cell technologies can be broadly classified into analysis of either DNA (genomics, epigenomics) or RNA (transcriptomics) with newer applications around the corner moving to combine both within the same cell.
Single-Cell genomics
Of relevance to cancer biology, is the ability to study genetic variations in individual cells. Although bulk DNA sequencing (DNA-seq) can be used to infer clonal sub-populations based on variant allele frequency analysis, it cannot be used to definitively test the co-occurrence of specific mutations in individual cells. Thus, single-cell DNA sequencing (scDNA-seq) can reveal cancer clonal architecture in far greater detail.
Single-Cell Transcriptomics
Single-cell transcriptomics has been used to study cancer stem cells, metastasis-initiating cells, chemotherapy resistance, and cancer immune responses.
Single-Cell Epigenomics
Many epigenetic processes (including DNA methylation, histone modifications, and chromatin accessibility) become dysregulated in cancer, and this fact has been exploited in various clinical applications.Single cell epigenomics can reveal the regulatory processes that lead to transcriptional heterogeneity in cancer, with important clinical implications. Single-cell DNA methylation analysis has also been applied to characterize circulating tumor cells and response to epigenetic therapies.
Single-Cell multiomics
It is also possible to combine analysis of the genome, transcriptome, epigenome, and other modalities using single-cell multiomic analyses. These combinatorial approaches allow genetic regulation to be studied in incredible detail and were named the 2019 “Method of the Year” by Nature Methods.
Single-Cell and CRISPR
While the human genome was sequenced 20 years ago, we still don’t know the cellular function of most genes. Single-cell CRISPR screens are a great way to cluster genetic perturbations by phenotypes like differentiation, chromosomal instability, the cell cycle, retrovirus activation, alternative-polyadenylation, etc., not to mention the potential for combining scRNA-seq with other phenotypes like imaging or protein measurements. By understanding mechanisms, it should become easier to rationally target multiple genetic dependencies in cancer.
Spatial Transcriptomics
Spatial single-cell transcriptomics is the next wave after single-cell analysis and will be particularly useful to labs studying human disease. Spatial transcriptomics, is the Nature’s 2020 “Method of the Year” and it can be performed on tissue sections using barcode arrays that record the coordinates of mRNA molecules in a sample. This technology was first applied in prostate cancer studies.
Spatial techniques can be divided into those that involve gene expression analysis on micro dissected tissues and those that involve in situ hybridization, in situ sequencing, in situ capturing, and computational reconstruction of spatial data.
Single-molecule fluorescence in situ hybridization (smFISH) is “the beginning of the hybridization-based approaches” with spatial techniques. In this method, multiple oligonucleotides carry fluorescent labels and bind to an RNA molecule. smFISH yields a quantitative mRNA readout with “a near 100% detection sensitivity”.
Single-Cell proteomics
Recent studies have used high-sensitivity mass spectrometry to achieve single-cell proteomics,and another report has coupled click chemistry with mass spectrometry to study lipid metabolism in single cells. Thus, it will soon be possible to study cell signaling pathways and altered metabolism in single cells.
Single-cell technologies are providing unique insights into disease biology and treatment response.
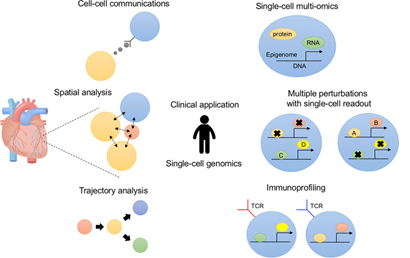
Figure 2: Overview of single-cell Genomics to understand disease pathogenesis. (Source of the figure: Seitaro Nomura, J Hum Genet, 2021)
The “cost per cell” currently remains prohibitive for routine analysis. However, it is to be expected that these costs will fall with time as they did for bulk sequencing, allowing this technology to eventually be used in routine patient care. Perhaps it does seem impossible, overwhelming, or ambitious to talk of these numbers, but that’s what was said about the human genome project over 20 years ago.
Pushing single-cell sequencing into clinical application is one of the important missions for clinical and translational medicine (CTM), although there still are a large number of challenges to be overcome.
References
1. Single‐cell RNA sequencing technologies and applications: A brief overview, Dragomirka Jovic et.al, Clin Transl Med, 2022 Mar,12
Emerging single-cell technologies have provided us with a powerful tool to dissect the clonal complexity of tumor cells, deconvolute the role of immune cell types in disease mechanisms, and monitor risk and treatment strategies to guide early patient diagnosis, since being highlighted as the ‘method of the year’ in 2013. As our capabilities in single cell sequencing continue to increase, latest advances in multi-omics of single cells are providing newer ways of integrating single cell transcriptomics with the multiple molecular measurements in a single experiment.
By Savita Jayaram Ph. D., Sheethal Umesh Nagalakshmi, Anay Limaye, Kushal Suryamohan Ph. D. , MedGenome Scientific Affairs
Emerging single-cell technologies have provided us with a powerful tool to dissect the clonal complexity of tumor cells, deconvolute the role of immune cell types in disease mechanisms, and monitor risk and treatment strategies to guide early patient diagnosis, since being highlighted as the ‘method of the year’ in 2013. As our capabilities in single cell sequencing continue to increase, latest advances in multi-omics of single cells are providing newer ways of integrating single cell transcriptomics with the multiple molecular measurements in a single experiment.
MedGenome provides novel assay and bioinformatics services to analyze multimodal single-cell datasets such as: CITEseq that simultaneously interrogates RNA and surface protein expression in single cells via the sequencing of antibody-derived tags (ADTs) and ATACseq that leverages transcriptome changes alongside chromatin accessibility and nucleosome occupancy. Concurrent estimation of both protein and transcript levels opens opportunities to use CITE-Seq in various biological areas, for instance, to profile disease heterogeneity, identifying rare cell sub-populations and novel subtypes, and to explore the mechanisms of host-pathogen interactions. ATACseq assays, on the other hand, can be applied to investigate chromatin accessibility signatures in diseases like macular degeneration and in human cancers, mapping transcription factor binding sites, exploring disease-relevant gene regulation, and studying evolutionary divergence of enhancer regions during development. Additionally, single-cell data can be used to reconstruct lineage trajectory maps, that can enhance our understanding of cell-fate transitions and identify putative branch points. Spatial transcriptomics provide users with extra insights into the cellular biology by providing a three-dimensional spatial context at single-cell resolution, and can be applied to both FFPE and frozen tissue sections. We have handled several such ‘multiome’ projects that have required customization/optimization of the lab protocols which helped us better understand the various QC checkpoints from both a wet lab and an analysis perspective. We have streamlined appropriate protocols, and built robust analysis pipelines, incorporating the latest tools and workflows.
Multimodal Analysis Workflow:
Although, single-cell transcriptomics has transformed our ability to characterize cell states, deep biological understanding requires advanced workflows such as the one depicted in the schematic below. A key analytical challenge is to integrate these multiple modalities to better understand cellular identity and function.1 Single-cell analysis tools need to accommodate different levels of resolution and throughput of the different datatypes, to comprehensively analyze the single cells at molecular level.
Figure 1: Multimodal Workflow
Single Cell CITEseq Analysis:
CITE-Seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing) is a multimodal single cell phenotyping method for performing RNA sequencing along with gaining quantitative and qualitative information on surface proteins with available antibodies on a single cell level.2 CITE-seq uses DNA-barcoded antibodies to convert detection of proteins into a quantitative and ‘sequenceable’ readout. Antibody-bound oligos act as synthetic transcripts that are captured during most large-scale oligodT-based scRNA-seq library preparation protocols (for e. g. 10x Genomics, Drop-seq, ddSeq). This allows for immunophenotyping of cells with a potentially limitless number of markers and unbiased transcriptome analysis using existing single-cell sequencing approaches. For phenotyping, this method has been shown to be as accurate as flow cytometry which is considered as the gold standard for absolute quantitative measurements. It is currently one of the main methods, to evaluate both gene expression and protein levels simultaneously in different species. Recently, this method has been successfully applied to understand the ongoing immune response in COVID-19 patients with varying severity, revealing discrete cellular compartments that can be targeted for therapy.3 The single-cell readout of both protein and transcript data at the same time can uncover novel information on protein-RNA correlations enabling precision health assessments. The increased copy number of protein molecules compared to RNA molecules typically leads to more robust detection of protein features. The protein data in CITE-seq may therefore represent the most informative modality.2 For data analysis, we leverage the weighted nearest neighbor (WNN) analysis provided by Satija lab in Seurat R package, which is an unsupervised strategy that defines the cellular state based on a weighted combination of both modalities.2 We find the WNN algorithm successfully recapitulates the biological expectations in comparison to separate analysis of each modality where certain cell populations can be masked allowing one datatype to compensate for weaknesses in another, demonstrating the importance of joint analysis. Additionally, this methodology enables interpretation of sources of heterogeneity from single-cell transcriptomic measurements, and integration of diverse types of single-cell data.
Single Cell Multiome (ATACseq) Analysis:
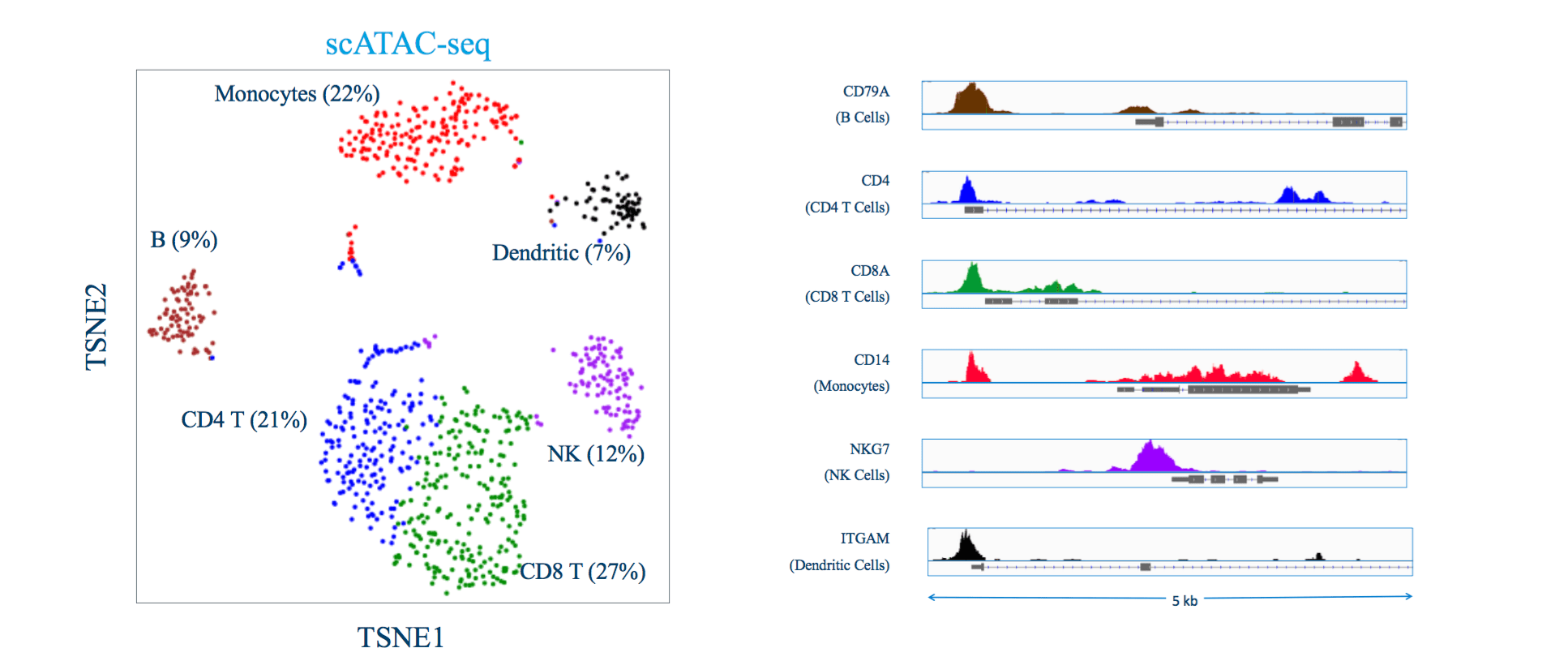
ATAC-seq aims at identifying DNA sequences located in open chromatin, i.e., genomic regions whose chromatin is not densely packaged and that can be more easily accessed by proteins than closed chromatin.4 The ATAC-seq technique makes use of an optimized hyperactive Tn5 transposase that fragments and tags the genome with sequencing adapters in regions of open chromatin. The output of the experiment is millions of DNA fragments that can be sequenced and mapped to the genome of origin for identification of regions where sequencing reads concentrate and form “peaks”. The hyperactivity of the Tn5 transposase makes the ATACseq protocol a simple, time-efficient method that requires 500–50,000 cells. The major steps in ATAC-seq data analysis include (1) Quality control and alignment, (2) Peak calling, (3) Advanced analysis at the level of peaks, motifs, nucleosomes, and TF footprints, and (4) Integration with multiomics data to reconstruct regulatory networks.4 ScATAC-seq can be applied in multiple situations including clinical specimens and developmental biology to study the heterogenous cell populations at single-cell resolution. However, this analysis is particularly challenging, due to both the sparsity of genomic data collected at single-cell resolution, and the lack of interpretable gene markers in scRNA-seq data. Similar to CITEseq, WNN analysis of Seurat can be applied to ATACseq data and it shows an increased ability to resolve cell states through integrated multimodal clustering. Further, ATACseq data analysis uses the Signac package developed by Satija lab, for the analysis of chromatin datasets. The cells are annotated using ScSorter, proven to have a higher annotation efficiency even for marker genes expressed at low levels.
Figure 2: ATACseq clusters and peaks (source: 10X Genomics)
Trajectory (Lineage) Analysis:
Trajectory inference has greatly boosted single-cell RNA-seq research by enabling the study of active and longitudinal changes vital to the discovery of genes governing lineages in the trajectory, or differentially expressed between groups. The wealth of information in the transcriptome of thousands of single cells can provide a snapshot of the dynamic changes at different levels of transition that is used to infer complex trajectories. The Monocle3 R package uses the concept of pseudotime, to order cells along a lineage based on the distance along a trajectory from its root or progenitor cells. For instance, in case of blood cell lineages, hematopoietic stem cells can be selected as the root cells. Monocle3 tracks these gene expression changes as a function of pseudotime, allowing for cells to have a branched structure when there are multiple possible outcomes. It can accurately resolve complicated biological processes and heterogenous cell populations, by learning an explicit principal graph based on advanced machine learning techniques called “Reversed Graph Embedding” followed by clustering.5 Subsequently, one can identify genes that are differentially expressed between different states such as control and experiment, or along the trajectories as cells transition from one state to another during development, disease or cell differentiation. Alternately, the velocity graph depicted in Figure B, describes cellular trajectories using RNA velocity (Velocyto or scVelo) that don’t rely on root cells but model the transitions based abundance of transcribed pre‐mRNAs (unspliced) to mature mRNAs (spliced).6 This can be easily identified in standard single‐cell RNA‐seq protocols due to the presence of introns, using Velocyto or loompy/kallisto counting pipeline.
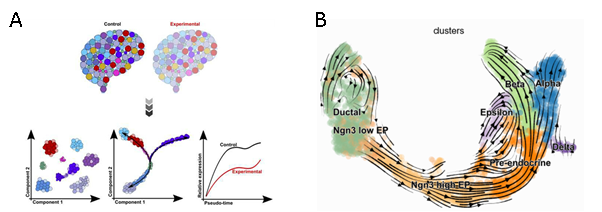
Figure 3: A) Figure showing a schematic from Ref. 7 of pseudotime trajectories examined between control and experiment systems using Monocle.7 B) Figure shows a velocity graph applied to endocrine development in the pancreas, with lineage commitment to four major fates: α, β, δ and ε-cells, with each arrow showing the direction and speed of movement of individual cells.
Spatial Transcriptomics:
Recent developments have sparked a growing interest in spatial transcriptomics technology coming from various platforms, such as, the Visium system from 10X Genomics utilizes spotted arrays of mRNA-capturing probes or SLIDEseq, a method developed at Harvard, for transferring RNA from tissue sections onto a surface covered in DNA-barcoded beads, with known positions. Nature Methods had crowned spatially resolved transcriptomics as the Method of the Year 2020.8 This method leverages spatial gene expression to identify genes and delineate neighbourhoods within fresh frozen or FFPE tissue sections. Using this approach, we can detect RNA species enriched in different subcellular compartments, observe distinct cell states corresponding to different cell-cycle phases, and reveal relationships between spatial position and molecular state. Each of these datasets represents an opportunity to understand principles governing the spatial localization of different genes in different cell types while capturing cellular boundaries (segmentations). Some of these methods use targeted panels i.e., they profile a pre-selected set of genes. Newer adaptations of Single-molecule FISH (smFISH) called as multiplexed error-robust FISH (MERFISH) can achieve near-genome-wide RNA profiling of spatially resolved individual cells with high accuracy and detection efficiency. The Seurat vignette for spatial data analysis uses SCTransform-based normalization, followed by dimensionality reduction and clustering like other multi-modal datasets. However, in addition to UMAP embedding, it overlays the clusters on the images of the tissue sections providing a spatial visualization. It offers additional features to zoom in and visualize individual molecules at a higher resolution. Once zoomed-in, one can also visualize individual cell boundaries as well in all visualizations.
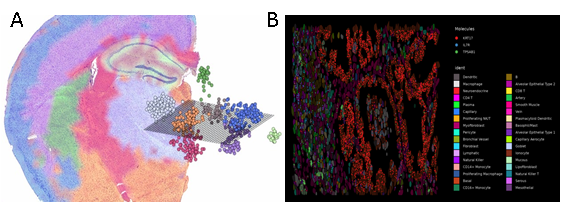
Figure 4: A) Figure shows how spatial transcriptomics provides a 2D or 3D visualization of transcriptomics data; B) Figure shows a zoomed in section of human lung tissue with visualization of cell types and expression localization patterns where individual cell boundaries can also be visualized (Seurat spatial vignette)
References
1. Stuart, T. et al. Comprehensive Integration of Single-Cell Data. Cell177, 1888-1902.e21 (2019).
2. Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell184, 3573-3587.e29 (2021).
3. Cambridge Institute of Therapeutic Immunology and Infectious Disease-National Institute of Health Research (CITIID-NIHR) COVID-19 BioResource Collaboration et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med.27, 904–916 (2021).
4. Yan, F., Powell, D. R., Curtis, D. J. & Wong, N. C. From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol.21, 22 (2020).
6. Bergen, V., Soldatov, R. A., Kharchenko, P. V. & Theis, F. J. RNA velocity—current challenges and future perspectives. Mol. Syst. Biol.17, e10282 (2021).
7. Kulkarni, A., Anderson, A. G., Merullo, D. P. & Konopka, G. Beyond bulk: A review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol.58, 129–136 (2019).
8. Marx, V. Method of the Year: spatially resolved transcriptomics. Nat. Methods18, 9–14 (2021).