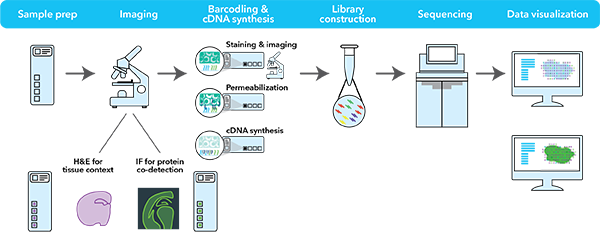
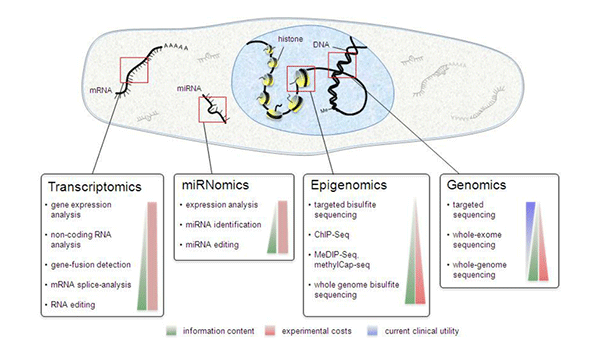
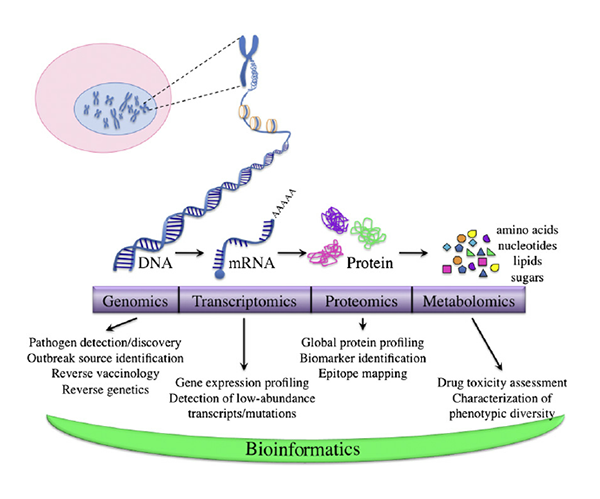
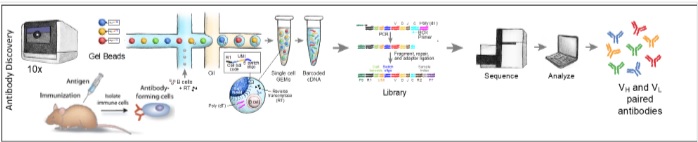
By Dr. Lavanya Balakrishnan and Vinay C. G., MedGenome Scientific Affairs
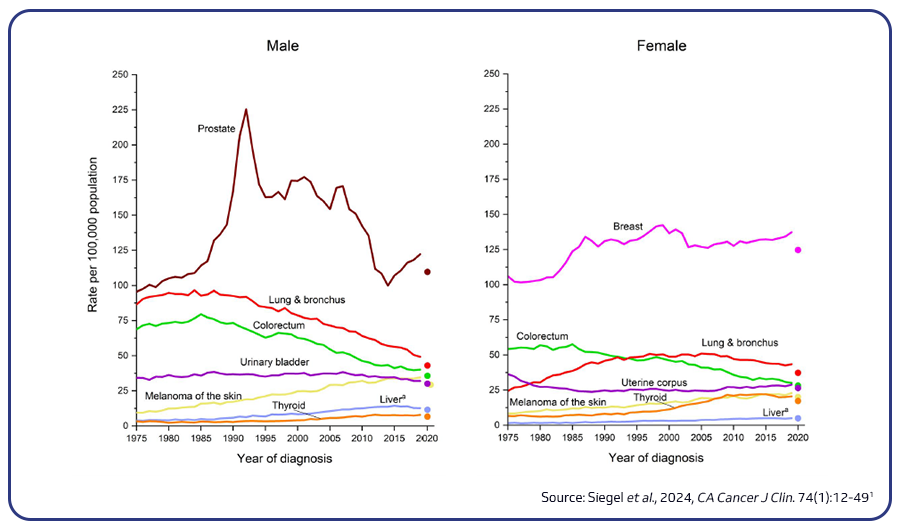
Lung cancer remains the leading cause of cancer-related deaths globally, accounting for 18.4% of all cancer fatalities. Smoking is the primary cause of lung cancer, responsible for 80-90% of lung cancer deaths, though other risk factors like secondhand smoke, radon, asbestos, and family history also play roles. Research efforts to improve early detection, such as low-dose CT scans and biomarker tests, offer promise, and machine learning tools are being developed to enhance CT scan accuracy1,2.
Classification and subtypes of lung cancer
Lung cancer is categorized into two main types: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC accounts for 80-85% of cases and includes subtypes like adenocarcinoma (LUAD), squamous cell carcinoma (LUSC), and large cell carcinoma (LCC). LUAD, the most common subtype, originates from mucus-producing lung cells and disproportionately affects non-smokers, especially women, constituting about 40% of NSCLC cases. LUSC, which makes up 20-30% of NSCLC, is strongly associated with smoking and tends to exhibit distinct mutation profiles. LCC, though less common (9-10%), is aggressive and poorly differentiated, often presenting as a large mass. SCLC is highly aggressive, with a five-year survival rate of only 5% among heavy smokers2.
Genetic drivers of lung cancer progression
Lung cancer is driven by diverse genetic alterations that affect its development, progression, and treatment response. In NSCLC, common driver mutations include EGFR, KRAS, MET, BRAF, ALK, ROS1, and RET. LUAD is frequently associated with mutations in tumor suppressor genes like TP53, STK11, and KEAP1, while LUSC tends to involve mutations in FGFR1 and AKT1. In SCLC, aggressive growth is driven by mutations in tumor suppressors such as TP53 and RB1, alongside the activation of MYC oncogenes, leading to rapid tumor progression and treatment resistance3.
Immunotherapy: Transforming lung cancer treatment
Lung cancer has traditionally been difficult to treat due to its high mutational burden and rapid progression, which often limit the effectiveness of conventional therapies like surgery, chemotherapy, and radiation. Targeted therapies have improved outcomes for some NSCLC cases with specific genetic mutations, but most individuals, including those with SCLC, still rely on platinum-based chemotherapy, which provides limited benefit.
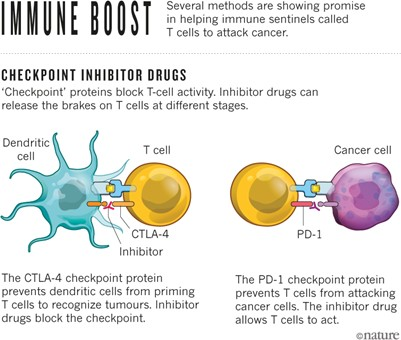
Among the various immune checkpoints that tumors exploit to evade the host immune system, the most well-known and clinically advanced are the programmed cell death protein-1 (PD-1)/programmed cell death ligand-1 (PD-L1) and cytotoxic T-lymphocyte antigen-4 (CTLA-4) pathways. The introduction of immune checkpoint inhibitors (ICIs) has revolutionized lung cancer treatment, particularly in NSCLC. ICIs like nivolumab, pembrolizumab, and atezolizumab block PD-1 and PD-L1, proteins that typically prevent T-cells from attacking cells. By releasing these “brakes,” ICIs empower T-cells to target and destroy cancer cells. Clinical trials have demonstrated the efficacy of these treatments. For instance, the KEYNOTE-024 trial, a landmark study, showed that pembrolizumab significantly improved overall survival in individuals with advanced NSCLC and high PD-L1 expression compared to standard chemotherapy. Additionally, the IMpower150 trial revealed that combining atezolizumab with chemotherapy and bevacizumab improved survival in advanced NSCLC cases, including those with lower PD-L1 expression. These breakthroughs have transformed the treatment landscape, offering substantial survival benefits, particularly in NSCLC cases without specific actionable mutations, though the impact in SCLC has been more modest, with only slight improvements when ICIs are combined with chemotherapy4.
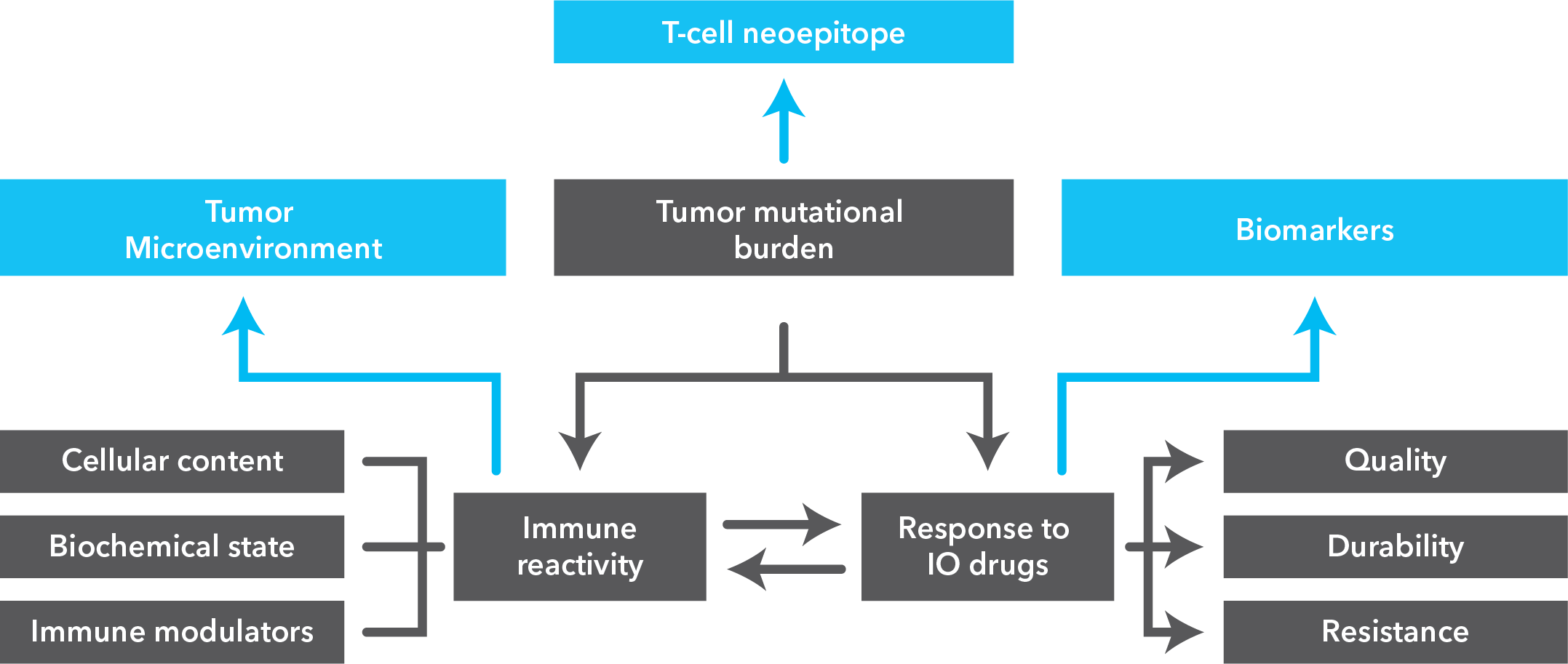
Role of biomarkers in immunotherapy response
Although immunotherapy has transformed the treatment landscape, not all individuals respond to ICIs, and some experience severe immune-related side effects. Biomarkers such as PD-L1 expression and tumor mutational burden (TMB) can help identify those who may respond favorably to immunotherapy. For instance, higher PD-L1 expression is often associated with better outcomes in metastatic NSCLC, while elevated TMB has been linked to improved responses in certain lung cancer subtypes. However, co-mutations, such as those involving STK11/LKB1, can predict poor outcomes, and individuals with EGFR mutations or ALK fusions generally show low response rates to ICIs5.
Circulating tumor DNA (ctDNA) has emerged as a promising biomarker for monitoring response to immunotherapy, though further validation is needed to establish its clinical utility. Recently, researchers have identified a set of 140 genes that may predict better outcomes in individuals with NSCLC undergoing immunotherapy combined with low-dose radiation, potentially offering new avenues for personalized treatment strategies6.
Single-cell sequencing in lung cancer immunotherapy
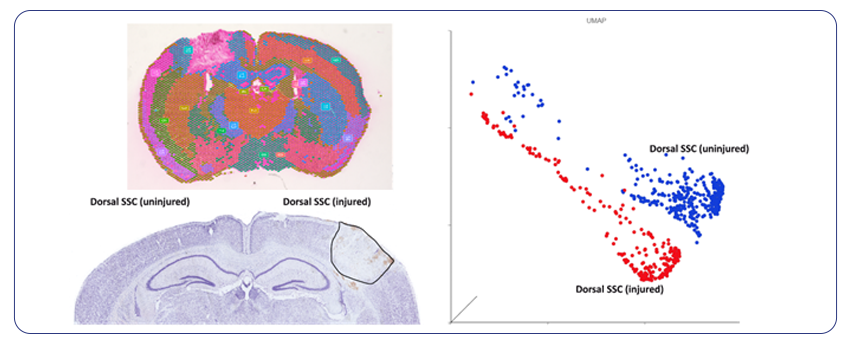
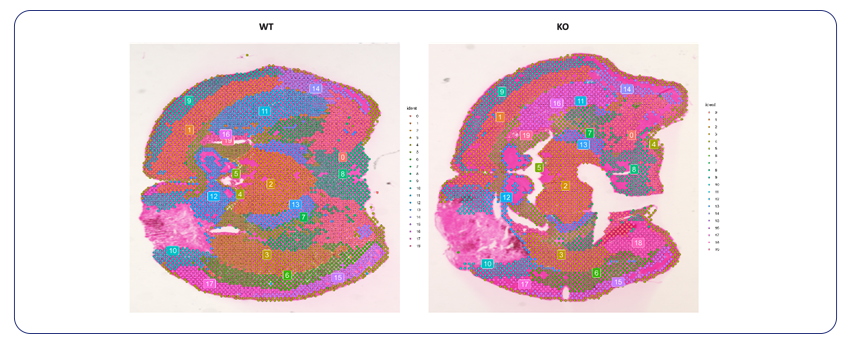
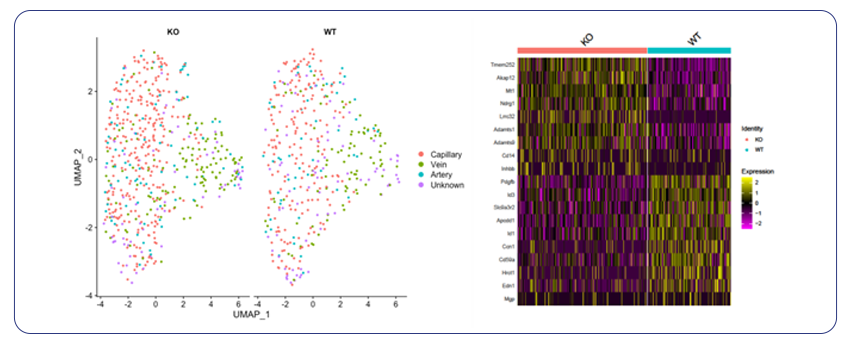
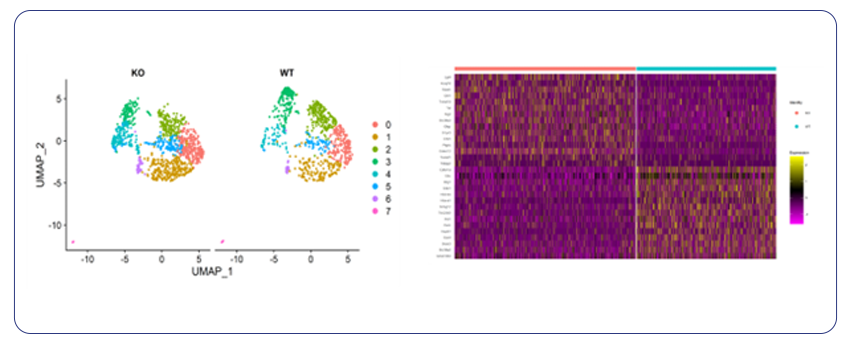
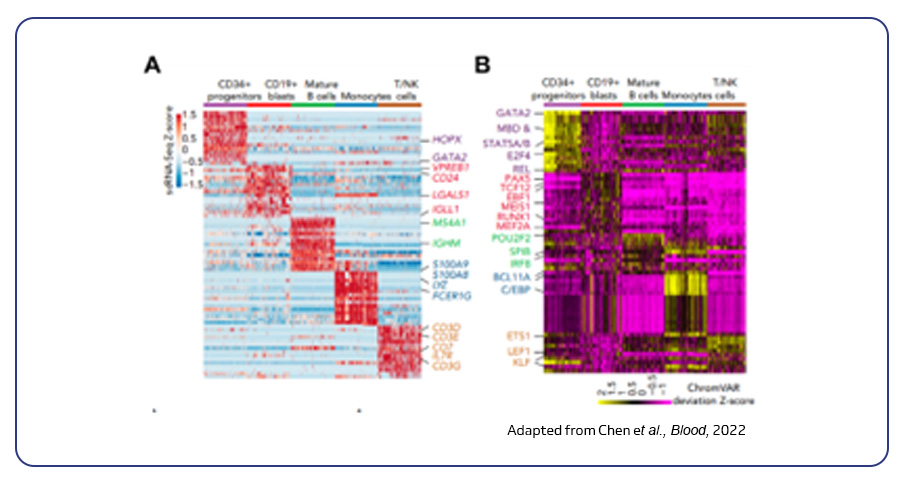
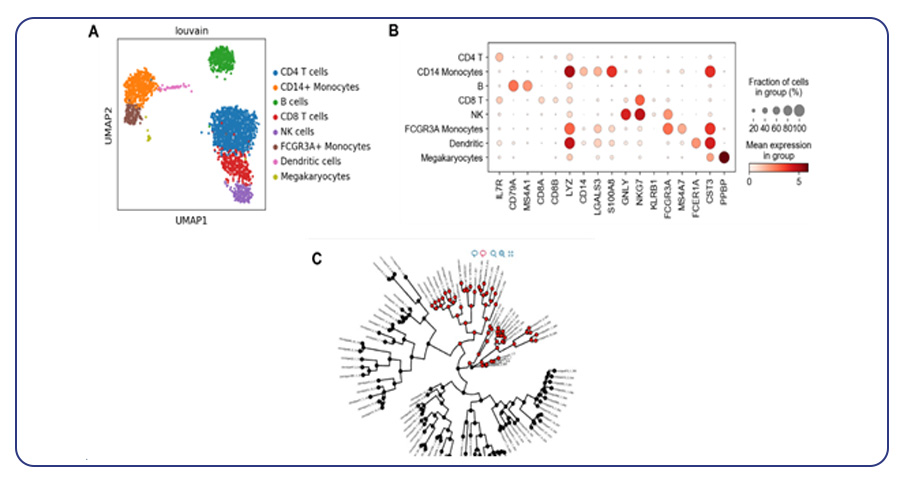
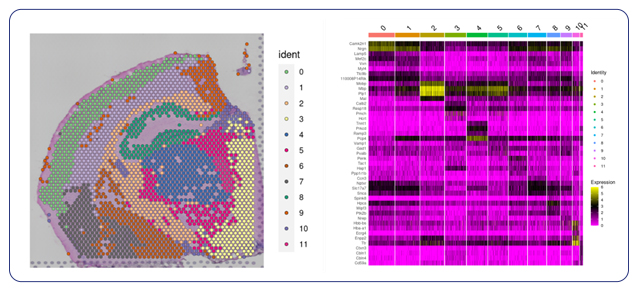
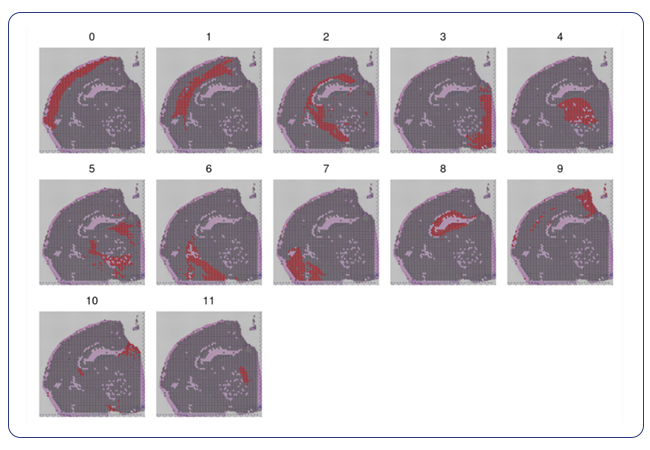
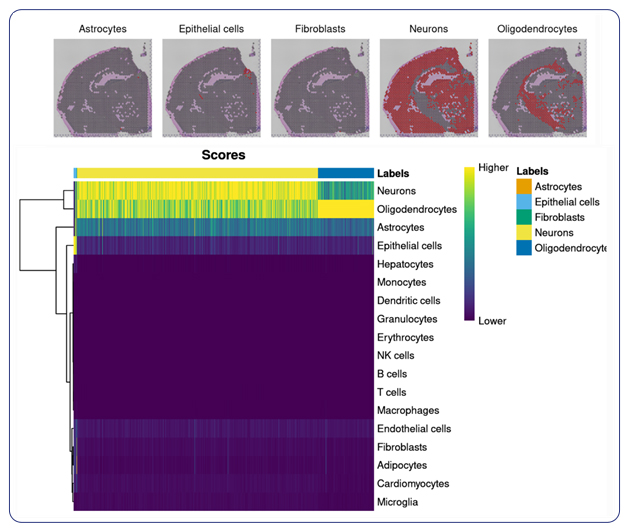
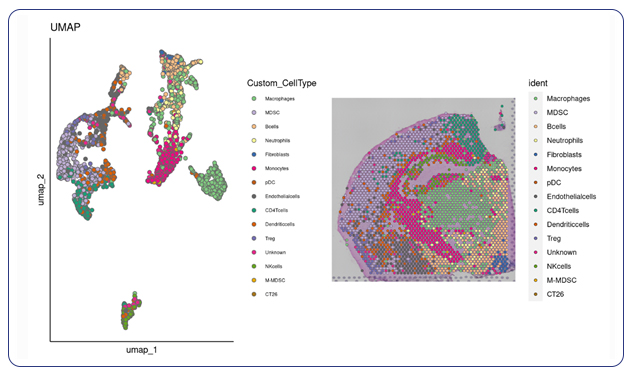
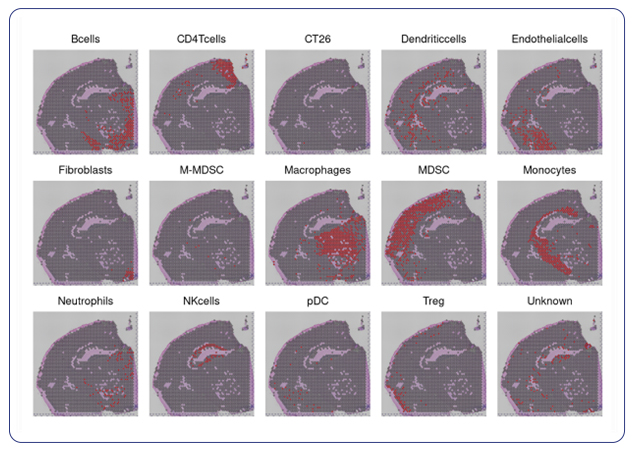
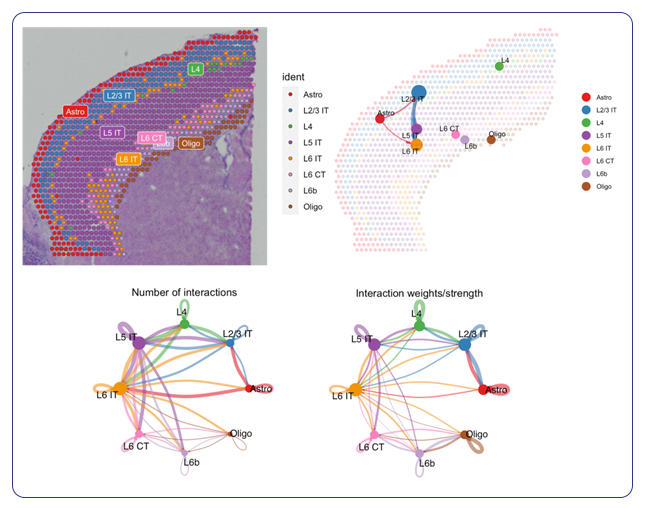
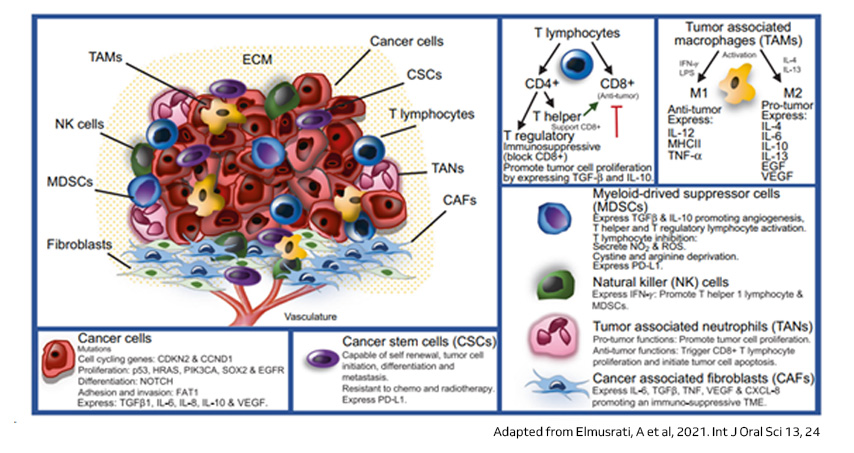
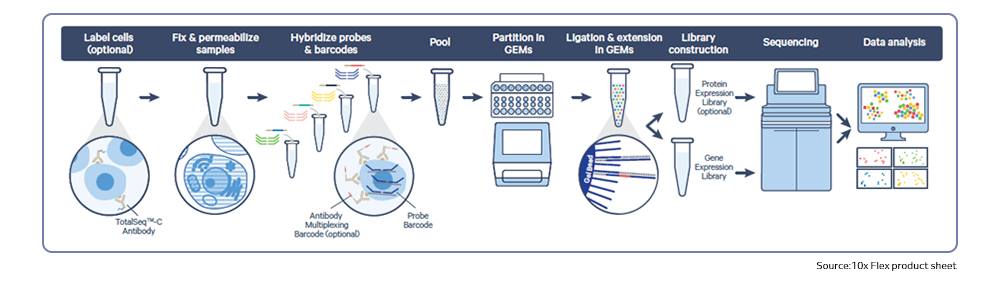
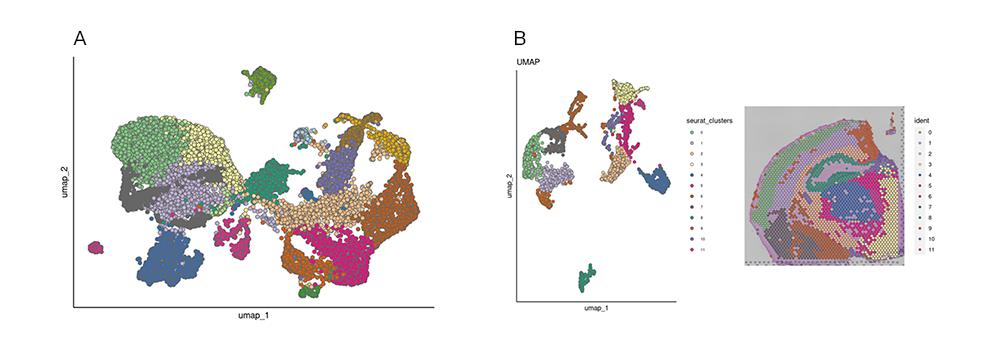
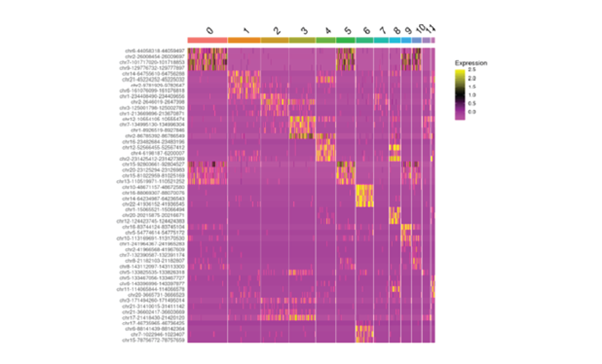
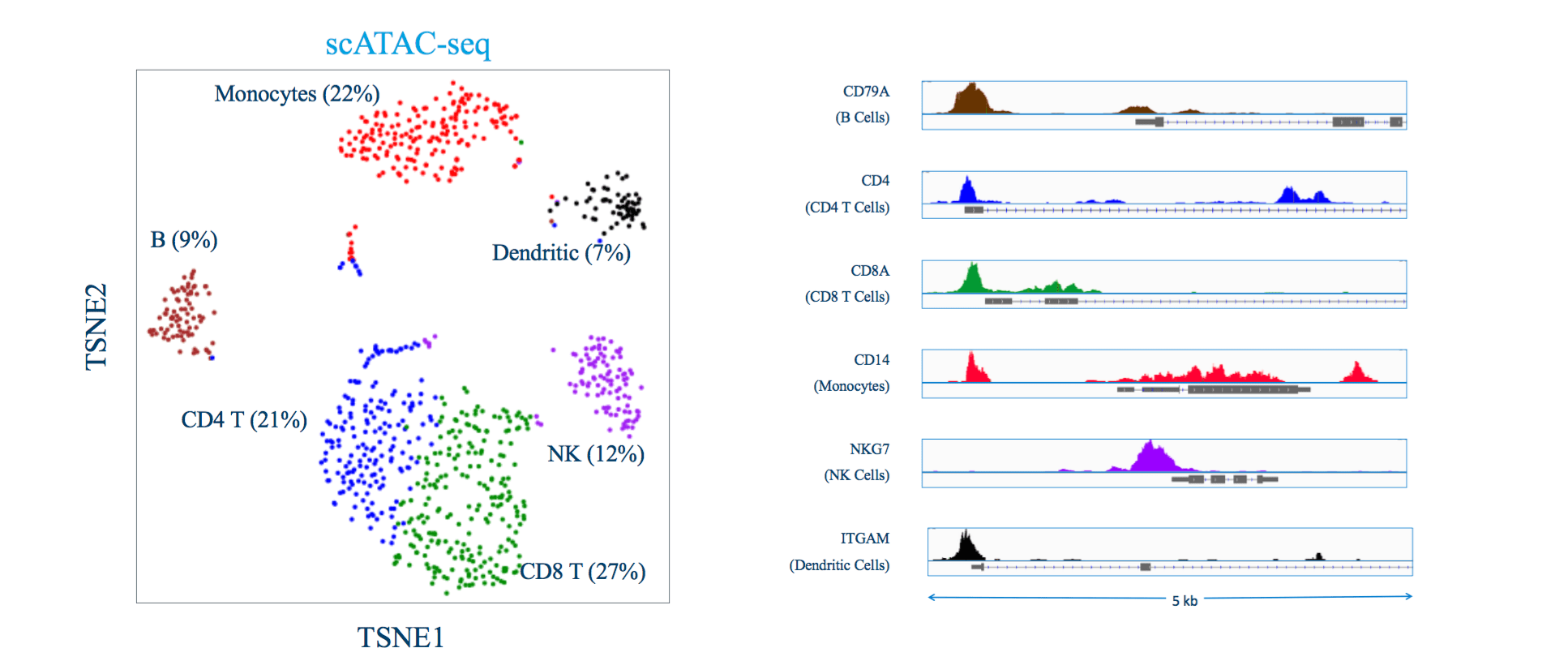
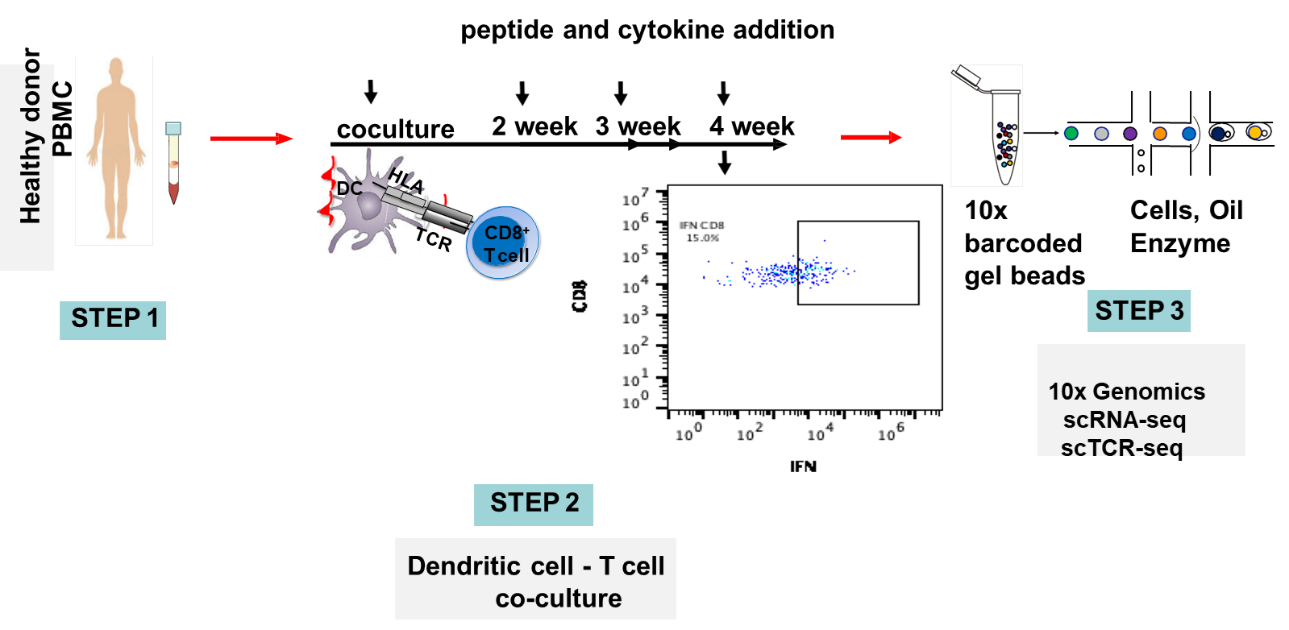
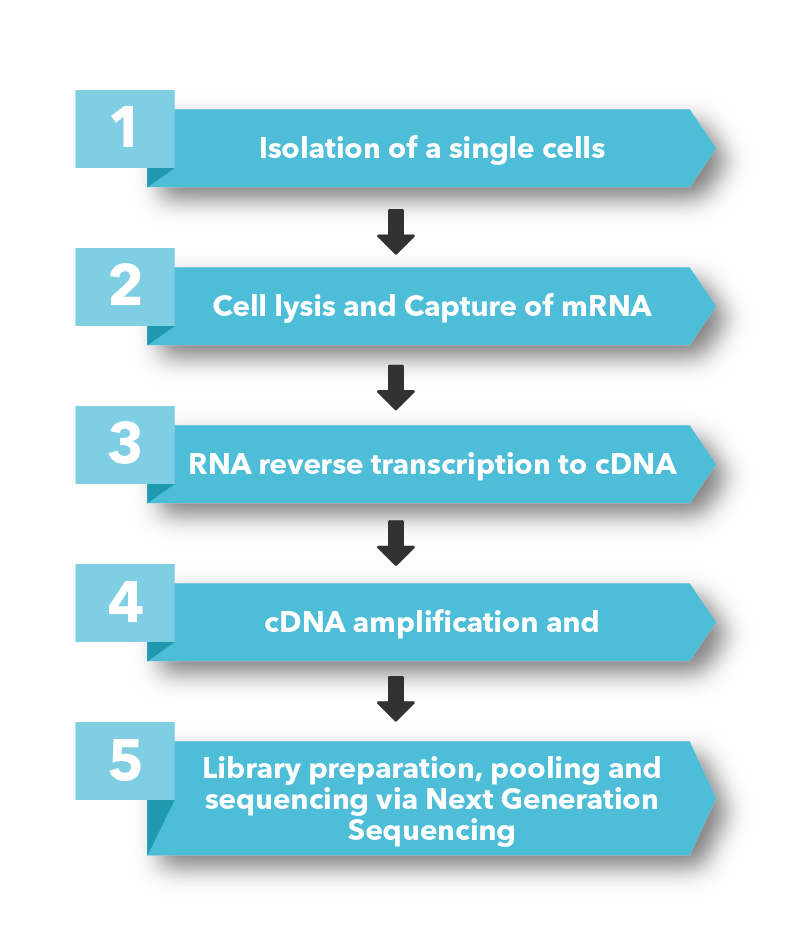
Single-cell sequencing has emerged as a pivotal tool for unraveling immunotherapy sensitivity in lung cancer. This advanced technology enables researchers to investigate the genetic, cellular, and molecular landscapes of tumors at a granular level, particularly in the tumor microenvironment (TME). By studying how different cells, including immune cells, interact within tumors, single-cell sequencing has uncovered mechanisms of immune regulation and resistance to therapies, especially immune checkpoint inhibitors (ICIs).
Research using single-cell RNA sequencing (scRNA-seq) has identified key immune cell populations in both non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), shedding light on the TME’s role in patient response to immunotherapy. Importantly, studies have linked low immune cell infiltration in the TME to better responses to ICIs, illustrating the impact of tumor heterogeneity on treatment resistance. Multi-regional sampling and sequencing have further mapped clonal evolution in lung tumors, helping predict resistance and improve treatment strategies7.
Additionally, single-cell sequencing has identified new biomarkers and therapeutic targets in lung cancer. For instance, KAT2B has shown promise as a predictive biomarker for response to immunotherapy in NSCLC8. This technology has also proven valuable in analyzing circulating tumor cells (CTCs) in liquid biopsies, uncovering critical biomarkers for lung cancer prognosis, metastasis, and treatment response, further advancing the development of personalized lung cancer therapies.
Conclusions
In conclusion, while lung cancer remains a leading cause of cancer-related deaths due to its aggressive nature and high mutational load, recent advances in immunotherapy, particularly immune checkpoint inhibitors, have significantly improved survival for many individuals, especially those with advanced NSCLC. However, variability in response highlights the need for precise biomarkers to guide treatment decisions. By integrating immunotherapy with advanced technologies like single-cell sequencing, future lung cancer treatments can be more personalized, improving outcomes.
MedGenome’s comprehensive genomic solutions for lung cancer research
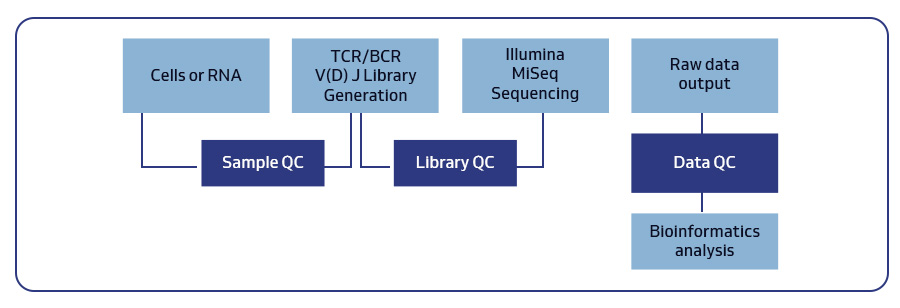
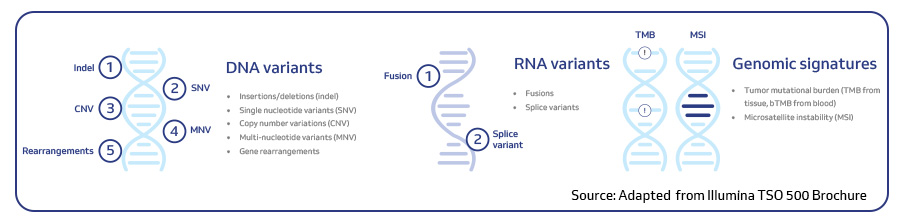
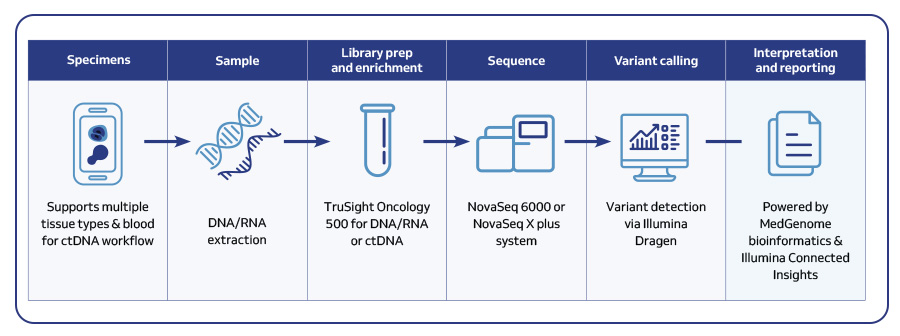
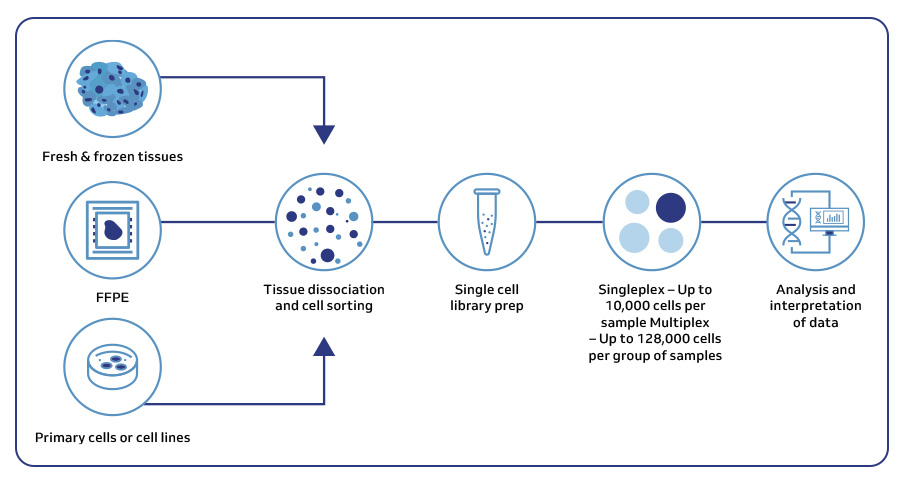
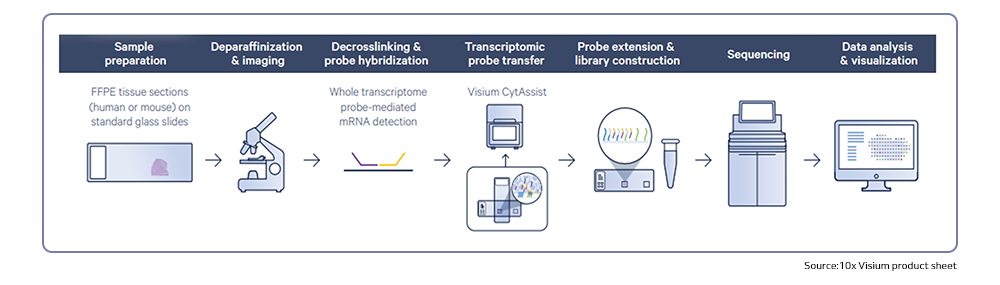
MedGenome offers advanced NGS solutions to accelerate lung cancer research, specializing in streamlined workflows and single-cell sequencing as a certified 10x Genomics provider. Our services include expert consultation, efficient sample processing, rapid data delivery, and customized visualizations. We leverage Illumina TruSight Oncology 500 (TSO500) for comprehensive genomic profiling, identifying key biomarkers like microsatellite instability (MSI) and TMB, as well as detecting fusions, indels, SNVs, and amplifications. Our powerful bioinformatics platform transforms raw sequencing data into high-quality, actionable reports, driving faster breakthroughs in understanding lung cancer and guiding the development of precision therapies. Additionally, our proprietary algorithm, OncoPeptTUME™ maps the tumor microenvironment using RNA-seq data, offering insights into tumor heterogeneity and immune interactions, customizable for cancer immunotherapy research.
Please reach out to our expert scientific team at research@medgenome.com for any questions or further information.
To know more about our advanced genomics solutions and services please click on the following links: Spatial transcriptomics, Single cell sequencing, RNA sequencing, Immune profiling, Cancer panels, Whole genome and whole exome sequencing and Epigenetic profiling
References
-
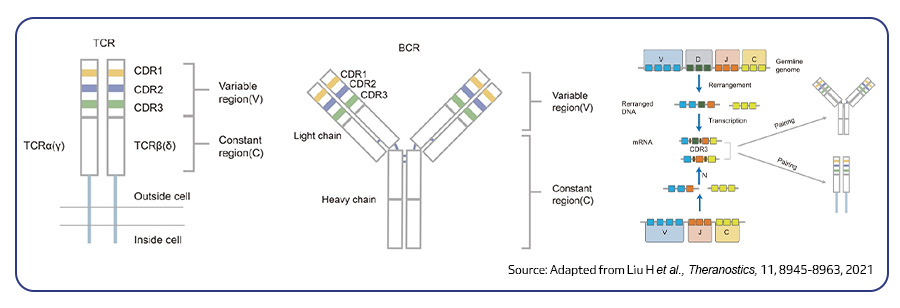
- Liu H, Pan W, Tang C, Tang Y, Wu H, et al. The methods and advances of adaptive immune receptors repertoire sequencing. Theranostics. 11, 8945-8963 (2021).
- Padinharayil H, Alappat RR, Joy LM, Anilkumar KV, Wilson CM, et al. Advances in the Lung Cancer Immunotherapy Approaches. Vaccines (Basel). 10, 1963 (2022).
- Lahiri A, Maji A, Potdar PD, Singh N, Parikh P, et al. Lung cancer immunotherapy: progress, pitfalls, and promises. Mol Cancer. 22, 40 (2023).
- Yan Y, Shen S, Li J, Su L, Wang B, et al. Cross-omics strategies and personalised options for lung cancer immunotherapy. Front Immunol. 15, 1471409 (2024).
- Mamdani H, Matosevic S, Khalid AB, Durm G, Jalal SI. Immunotherapy in Lung Cancer: Current Landscape and Future Directions. Front Immunol. 13, 823618 (2022).
- Altorki NK, Bhinder B, Borczuk AC, Elemento O, Mittal V, McGraw TE. A signature of enhanced proliferation associated with response and survival to anti-PD-L1 therapy in early-stage non-small cell lung cancer. Cell Rep Med. 5, 101438 (2024).
- Xiao N, Liu H, Zhang C, Chen H, Li Y, et al. Applications of single-cell analysis in immunotherapy for lung cancer: Current progress, new challenges and expectations. J Adv Res. S2090-1232(24)00462-4 (2024).
- Zhou X, Wang N, Zhang Y, Yu H, Wu Q. KAT2B is an immune infiltration-associated biomarker predicting prognosis and response to immunotherapy in non-small cell lung cancer. Invest New Drugs. 40, 43-57 (2022).
#Lung cancer, #Lung adenocarcinoma, #Non-small cell lung cancer (NSCLC), #Small cell lung cancer (SCLC), #EGFR, #ALK, #Immunotherapy, #Immune checkpoint inhibitors, #PD-L1, #Biomarkers, #tumor microenvironment, #Circulating tumor DNA, #Single cell RNA sequencing, #Tumor mutation burden, #TSO500